Inherited Cardiomyopathies: Genetics and Clinical Genetic Testing
2017-05-26GuoliangWang,RuiruiJi,WenxinZou等
Abbreviations
ARVD/C – arrhythmogenic right ventricular dysplasia/ cardiomyopathy
DCM – dilated cardiomyopathy
HCM – hypertrophic cardiomyopathy
LVNC – left ventricular noncompaction
NGS – next generation sequencing
RCM – restrictive cardiomyopathy
Introduction
Inherited cardiomyopathies are a group of cardiovascular disorders and a major cause of heart disease in all age groups, often with an onset in adolescence or early adult life. Based on ventricular morphological and functional features, inherited cardiomyopathies are classified into hypertrophic cardiomyopathy(HCM), dilated cardiomyopathy (DCM), arrhythmogenic right ventricular dysplasia/cardiomyopathy(ARVD/C), left ventricular noncompaction (LVNC),and restrictive cardiomyopathy (RCM) [1]. Since a mutation in the β-myosin heavy chain gene (MYH7)was first identified as being responsible for causing HCM [2], many advances have been made to define the genetic etiology of inherited cardiomyopathies.For instance, HCM is now viewed as a “disease of the sarcomere,” as most of the genes associated with HCM encode proteins of the myofilaments or Z-disc of the sarcomeres [3]. In contrast, DCM is far more genetically heterogeneous, with mutations in genes encoding cytoskeletal, nucleoskeletal, mitochondrial, and calcium-handling proteins [4]. Many genes have been identified for causing ARVC, most resulting in disturbed desmosome/intercalated disk function. Although understanding the genetic basis for the development of RCM and LVNC has been more elusive, genes for both entities have been identified and appear to include sarcomere dysfunction as a critical factor [5]. T he advent of next-generation sequencing (NGS) technologies has led to increasingly comprehensive testing for inherited cardiomyopathies. Understanding the genetic etiology for these disorders has improved their clinical recognition and management and led to new guidelines for treatment and family-based diagnosis and surveillance[6]. In this review, we introduce genetics and clinical genetic testing for inherited cardiomyopathies.
Hypertrophic Cardiomyopathy
HCM (HCM, OMIM: 612098) is characterized by asymmetric or concentric wall thickening in the absence of an underlying systemic condition or other cardiac disease. With an estimated prevalence of 1 in 500 in the general population [7], HCM is the most common inherited heart condition. Although the age at onset of HCM can range from infancy to old age,manifestations usually do not appear before adolescence in carriers of a pathogenic variant. HCM is inherited primarily in an autosomal-dominant pattern,although reduced penetrance and clinical variability are common [8]. Clinically, most patients with HCM are asymptomatic or mildly symptomatic [9, 10]. The major effects of this disorder on human health are its predilection to be inherited, its reputation as the most common cause of sudden cardiac death (SCD) in young, healthy individuals, and its potential to develop heart failure (HF) because of diastolic factors or development of systolic dysfunction [5].
Genetics of Hypertrophic Cardiomyopathy
Since the pathogenic missense mutation in theMYH7(MYH7R403Q) was revealed two decades ago, hundreds of mutations have been identified in at least 44 putative HCM-susceptibility genes(Table 1) [2, 5, 11]. The most common genetic subtypes of HCM are sarcomeric- or myo filament-HCM, caused by mutations in eight genes encoding proteins of the myo filaments of the cardiac sarcomere [11]. In those patients with positive genetic tests, myosin-binding protein C (MYBPC3) andMYH7are, by far, the two most commonly identified HCM-associated genes, with an estimated prevalence of 25–35% for each. Other genes, includingTNNT2,TNNI3,TPM1,and ACTN2, are known to account each for a small proportion of patients (1 to 5%) [12]. Collectively, sarcomere variants are identified in as many as 60% of patients with HCM who also have a family history and in 40% of patients with sporadic HCM [13]. In addition to defects in the sarcomere-encoding genes, patients with HCM have been identified hosting mutations in Z-disk and other nonsarcomere-encoding genes. The giant protein titin (TTN) and its interactive Z-disc proteins, including muscle LIM protein (MLP), Z-band alternatively spliced PDZ-motif (ZASP), telethonin,nexilin, myopalladin, myozenin-2, α-actinin 2, cardiac ankyrin repeat protein (CARP), and vinculin,have been identified as causes of HCM when the respective gene is mutated [14]. Although almost 1000 variants for HCM have been identified to date,most are private and can, therefore, be detected only through comprehensive genetic testing [13].
Mutations identified in sarcomeric genes typically are single nucleotide substitutions and, in most instances, the mutant protein is thought to incorporate into the sarcomere and act in a dominant-negative manner. However, about a half of the reportedMYBPC3mutations are truncations caused by nonsense and frameshift mutations;these and someMYBPC3missense mutations can result in haploinsufficiency, a condition in which the gene product of the wild-type allele cannot compensate for the decreased product from the mutant allele [15]. For Z-disk and calcium modulator genes, the specific mechanism has not been clearly elucidated.
Dilated Cardiomyopathy
DCM (DCM, OMIM: 613694) is characterized by left ventricular dilation and systolic dysfunction (a
reduction in myocardial force generation) and is the most common indication for cardiac transplantation[1]. The annual incidence is 2–8/10,000 (0.57/100,000/year in children), and the estimated prevalence is 1/2500 population, although this fi gure may be underestimated [1, 16, 17]. The age at onset includes newborn through late adulthood, but most patients are diagnosed between 20 and 50 years of age [18]. Clinical manifestations include HF, thromboembolism, and SCD. DCM can also present with muscular involvement and may be the presenting or primary clinical feature of several multisystem conditions, including Emery-Dreifuss muscular dystrophy (EDMD), Barth syndrome, myofibrillar myopathy, limb-girdle muscular dystrophy (LGMD), and Duchenne or Becker muscular dystrophy (DMD/BMD) [19].
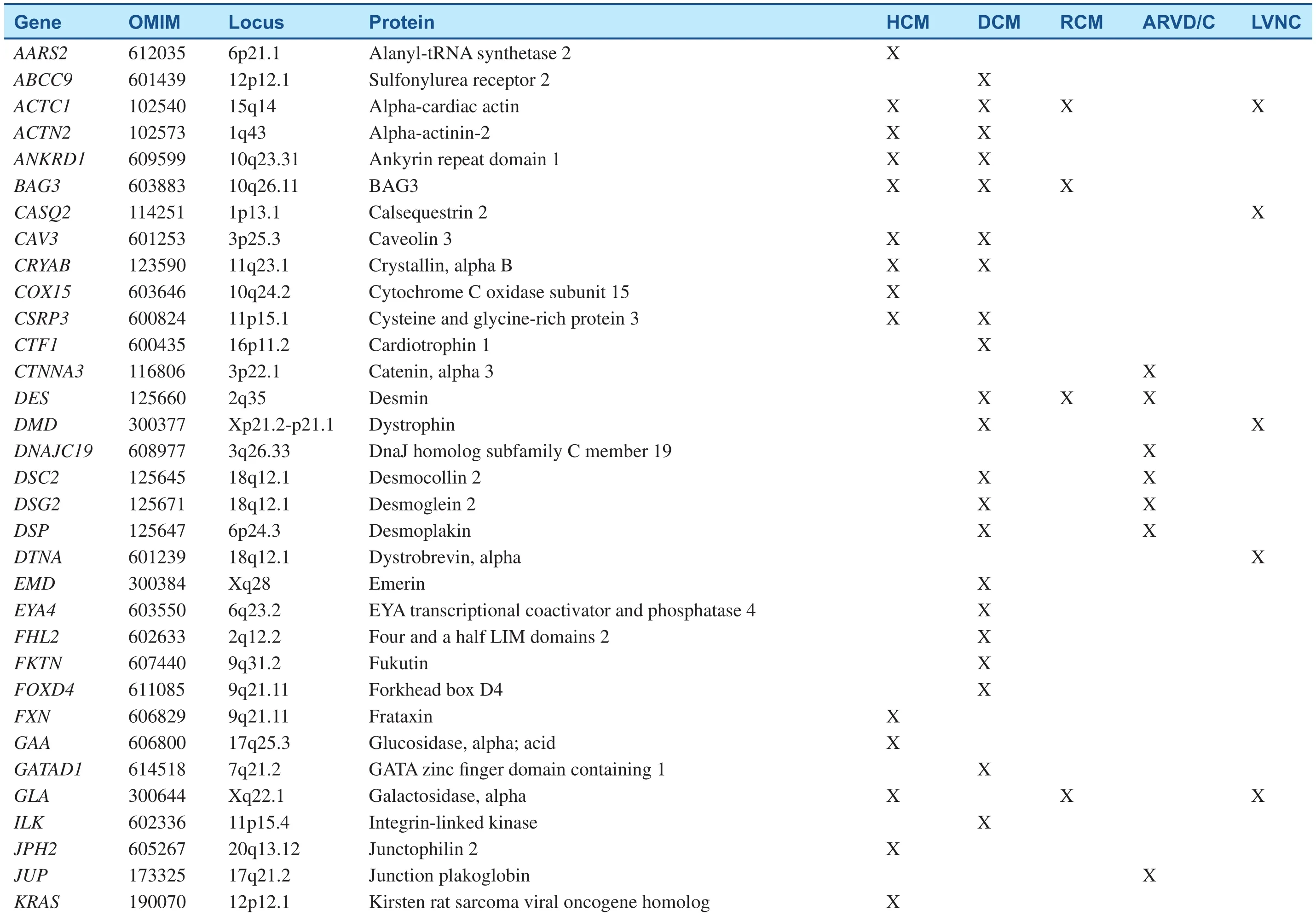
Table 1Inherited Cardiomyopathy-Associated Genes.
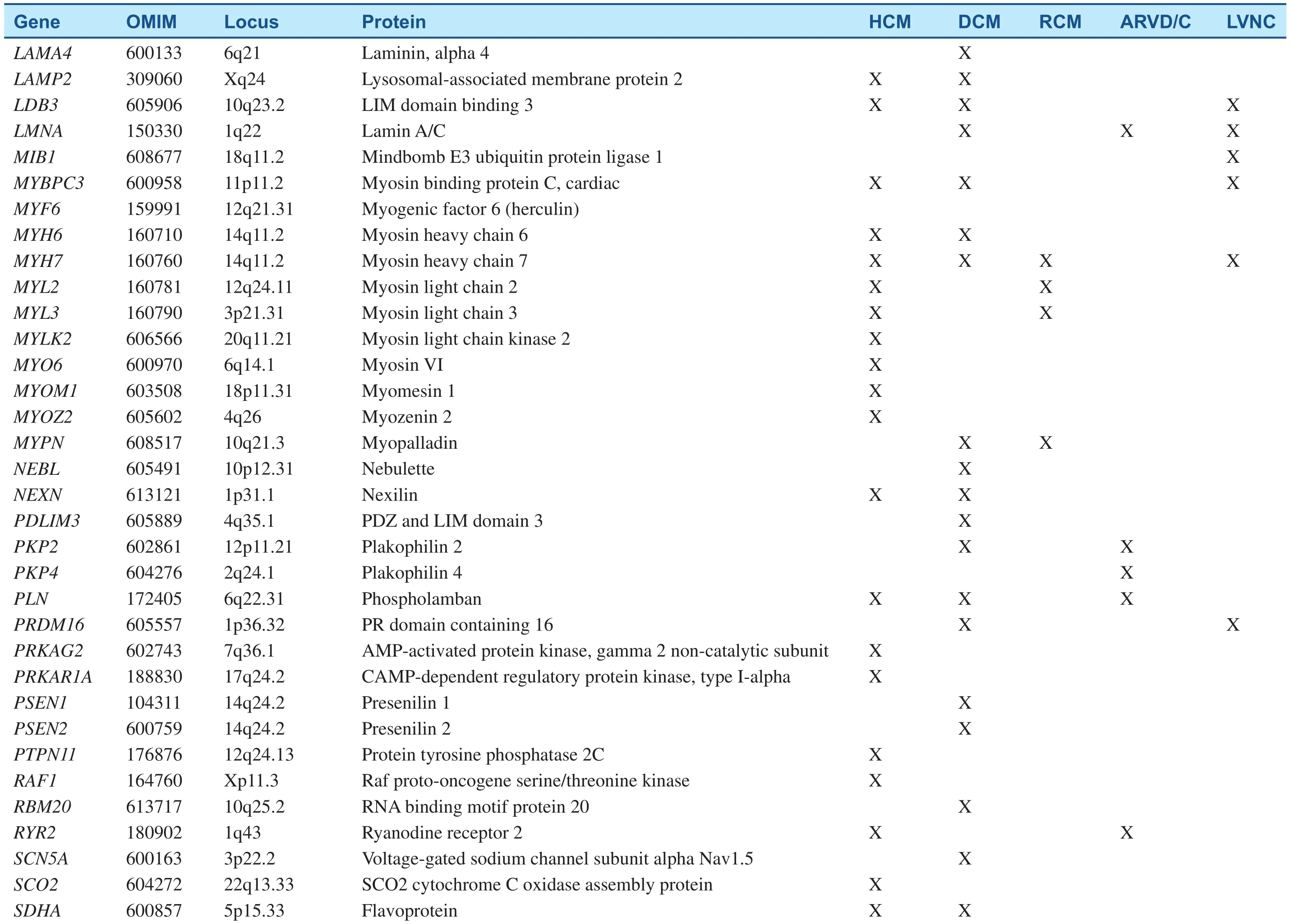
Table 1(Continued)
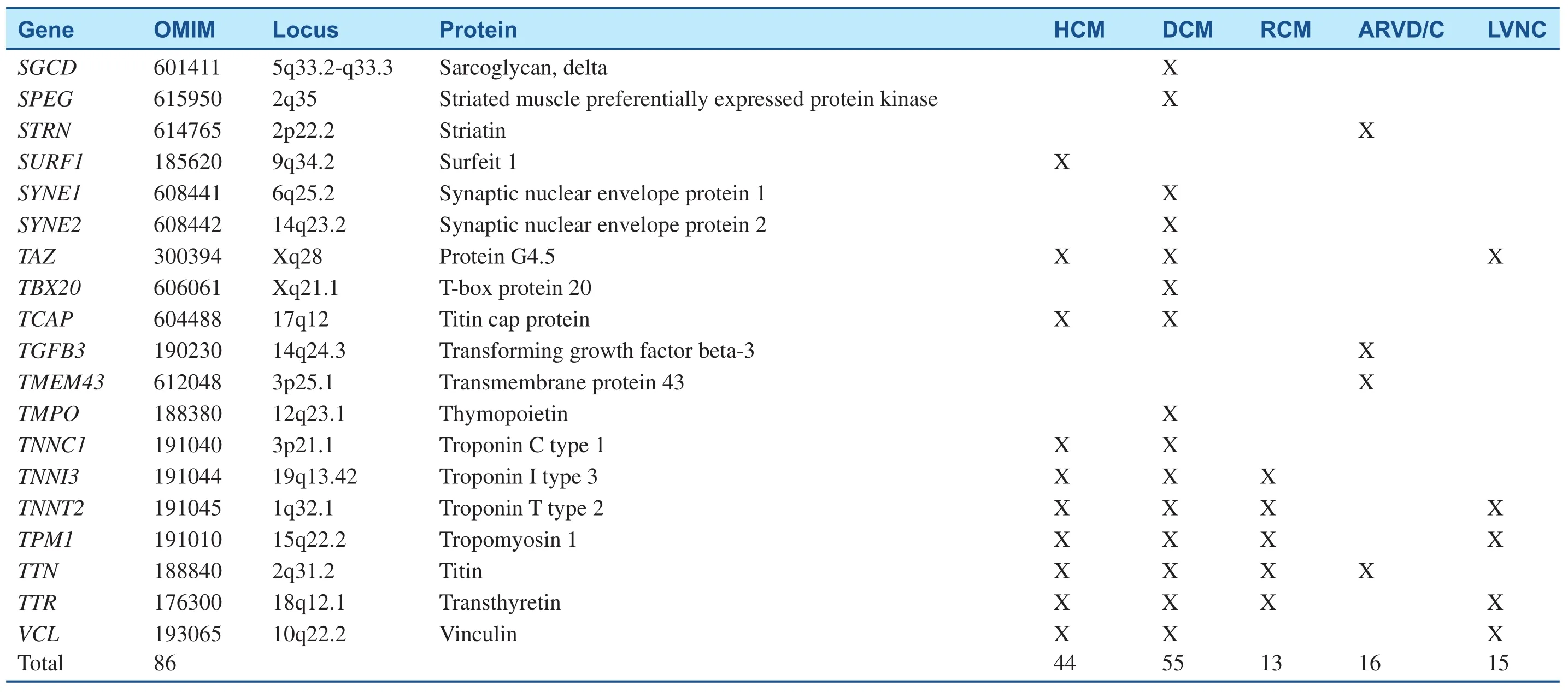
Table 1(Continued)
Genetics of Dilated Cardiomyopathy
DCM cases with a genetic etiology represent approximately 30–50%, based on the presence of a family history. More than 50 disease genes have been identified (Table 1); the most common mode of inheritance is autosomal-dominant transmission,although X-linked, autosomal-recessive, and mitochondrial inheritance forms have been described[4]. Most of these genes encode cytoskeletal,sarcomeric, or Z-disk proteins, but mutations in a small number of ion channel-encoding and desmosome-encoding genes also have been identified.They include genes primarily encoding cytoskeletal δ-sarcoglycan (SGCD), β-sarcoglycan (SGCB),desmin (DES), lamin A/C (LMNA), vinculin, sarcomeric/myo fibrillar (α-cardiac actin [ACTC],troponin T [TNNT2], troponin I [TNNI3]),MYH7, myosin-binding protein C, α-tropomyosin(TPM1), and Z-disk proteins MLP/ cysteine and glycine-rich protein 3 (CSRP3), TTN, telethonin/TCAP, α-actinin-2 (ACTN2), nebulette (NEBL),myopalladin (MYPN), ANKRD1/CARP, and ZASP/LIM- domain binding 3 (LBD3)). Ion channel encoding genes identified to date include the cardiac sodium channel geneSCN5Aand calcium homeostasis regulator phospholamban (PLN); and associated desmosome-encoding genes including desmoplakin (DSP), desmoglein-2 (DSG2),and desmocolin-2 (DSC2) have also been shown to result in a DCM phenotype [5]. New genes for DCM are continually being discovered, with recent additions including the gene encoding BCL2-associated athanogene 3 (BAG3) [20],RBM20 [21], and TTN [22]. TTN may contribute to as many as 25% of familial and 18% of sporadic DCM cases, rendering it by far the most commonly mutated gene in DCM [22].
Given the diversity of affected cellular processes,numerous proximal factors probably contribute to contractile dysfunction in DCM. Molecular mechanisms of DCM-causing mutations include diminished force generation and transmission, alterations of energy production and regulation, and intracellular calcium defects [23]. Sarcomere mutations may cause DCM through two mechanisms: deficits of force production and deficits of force transmission [24]. Mutations in cytoskeletal and Z-disc proteins cause mainly defects of force transmission.The altered desmosomal proteins appear to disrupt the links among the intercalated disk, Z-disk, and sarcomere [5]. Mitochondiral DNA mutations may alter energy production and regulation in affected cardiomyocyte [25], and PLN mutations cause intracellular calcium defects [26].
Restrictive Cardiomyopathy
Restrictive cardiomyopathy (RCM)(RCM1,OMIM# 115210; RCM3,OMOM#612422)is a rare disease of the myocardium characterized by increased stiffness of the ventricles leading to compromised diastolic filling with preserved systolic function. These changes may develop in association with local in flammatory or systemic,infiltrative, or storage disease [27]. RCM accounts for approximately 5% of all cases of primary heart muscle disease. In people with familial RCM, the heart muscle is stiff and cannot fully relax after each contraction. Most affected individuals have severe signs and symptoms of HF. Adult patients with RCM present with dyspnea, fatigue, and limited exercise capacity. In children, RCM may present with failure to thrive, fatigue and even syncope [28, 29]. RCM carries a poor prognosis,particularly in children, despite optimal medical treatment. Several studies have reported that 66–100% die or receive a cardiac transplant within a few years of diagnosis [30, 31].
Genetics of Restrictive Cardiomyopathy
RCM may be associated with systemic disease but is most often idiopathic. The results of recent molecular genetic investigations have revealed that a substantial proportion of RCM without associated systemic disease is caused by mutations in sarcomeric disease genes that have been associated with HCM, DCM, and noncompaction cardiomyopathy.Mutations in several genes have been found to cause familial RCM. Mutations in the cardiac troponin I gene (TNNI3) are the major causes of this condition.Mogensen et al., reported a large family in which individuals were affected by either idiopathic RCM or HCM. Linkage analysis to selected sarcomeric contractile protein genes identi fi edTNNI3as the likely disease-causing gene [27]. The fact thatTNNI3mutations were identified in a significant proportion of such patients indicates that idopathic RCM is part of the clinical expression of sarcomeric contractile protein disease and of HCM [32]. Chen et al. also reported aTNNI3missense mutation (R192H) in idiopathic RCM in a 12-year-old Chinese girl [33]. The case further improves the knowledge of the causes of cardiomyopathy disease and shows that the spectrum of sarcomeric gene mutations may be involved in pediatric RCM. Mutations in several sarcomeric genes have been reported subsequently in patients with RCM. Peddy et al., reported a novel presentation of a mutation in theTNNT2gene causing RCM in a child [34]. These findings, together with a possibly aggravating mutation inMYBPC3, further expand the phenotypic spectrum caused byTNNT2andMYBPC3mutations [34]. Mutations involving cardiac troponin T likely lead to altered calcium sensitivity of the troponin complex, contributing to altered relaxation of the cardiac muscle, which is the hallmark of RCM[34]. Ware et al. first described theMYH7mutation in a child with RCM. These findings expand the phenotypic presentation of mutations in this sarcomeric protein, previously well known to cause both HCM and DCM [35]. RCM can also be a manifestation of desmin-related cardiomyopathy, caused by mutations in theDES[36]. A point mutation inBAG3is known to cause fulminant skeletal myopathy and early death in knockout mice and myo fibrillar myopathies with RCM or HCM in humans [20]. Familial RCM also can be caused by a mutation inACTC1[31].
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C)
ARVD/C (ARVD/C9, OMIM# 609040) is a rare,inherited cardiomyopathy characterized by ventricular arrhythmias, SCD, and abnormalities of the right (and less commonly left) ventricular structure and function. It is an inherited condition with an estimated prevalence of 1 per 5000. Patients usually present during the second to fifth decades of life with palpitations, lightheadedness, syncope, or sudden death [37].
Genetics of ARVD/C
ARVD/C is inherited as an autosomal-dominant trait, meaning that the risk of a family member inheriting an abnormal gene is 50% for all offspring of the genetically affected proband, whether male or female [38]. Approximately 50–60% of patients with ARVD/C are estimated to have a mutation in genes associated with cardiac desmosomes [39].The desmosomal proteins involved are desmoplakin(encoded by theDSPgene), plakophilin 2 (PKP2),DSG2, DSC-2, and junctional plakoglobin (JUP).The nondesmosomal proteins related to ARVD/C are DES, transmembrane protein 43 (TMEM43),transforming growth factor β-3 (TGF β 3), LMNA,TTN, PLN, and α-T-catenin (CTNNA3). A mutation in the cardiac ryanodine receptor, encoded by the ryanodine gene (RyR2), was identified in a patient affected by ARVD/C [39]. To date, the majority of pathogenic mutations have been identified in genes coding for desmosomal proteins,with thePKP2gene being responsible for approximately 35–40% of cases. Mutations in the genesDSP,DSG2, andDSC2are responsible for nearly 15–20% of ARVD/C cases. The most prevalent form of ARVD/C (type 9) is caused by mutations in thePKP2gene, which encodes the PKP2, an essential armadillo repeat protein located in the outer dense plaque of cardiac desmosomes that interacts with numerous other cell adhesion proteins. How PKP2 mutations perturb cardiac desmosome assembly and function in ARVD/C is unknown. It has been speculated that lack of PKP2 or incorporation of mutant PKP2 into cardiac desmosomes impairs cell-cell contacts and, as a consequence, disrupts adjacent cardiomyocytes, particularly in response to mechanical stress or stretch. Mura et al. reported the first case of copy number variation (CNV)identified in an ARVC family using high-density single nucleotide polymorphisms (SNPs) arrays. A heterozygous deletion of about 122.5 Kb on chromosome 12p11.21, encompassing the entirePKP2gene, was detected in all affected family members.Hence,PKP2deletions may arise by non-recurrent rearrangements due to replication-based mechanisms of DNA repair [40].
Left Ventricular Noncompaction(LVNC)
LVNC (LVNC OMIM#604169), also known as spongy myocardium, is a distinct form of cardiomyopathy occurring in-utero when segments of spongy myocardium fail to transform into compact,mature musculature, resulting in prominent myocardial trabeculae, deep intra-trabecular recesses,and decreased cardiac function [41]. It was first described in 1990 [42]. Prevalance of LVNC is estimated to be approximately 0.25% of adults referred for echocardiography [43]. The clinical manifestation of LVNC is highly variable, ranging from no symptoms to a progressive deterioration in cardiac function that results in congestive HF, arrhythmias,thromboembolic events, and SCD [44].
Genetics of LVNC
LVNC is a genetically heterogeneous cardiomyopathy, with both familial and sporadic forms.Autosomal dominant is the most common form of inheritance, but autosomal recessive, X-linked, and maternally inherited (matrilineal) mitochondrial inheritance have been reported. More than ten genes have been described in LVNC, and some of the genetic mutations are associated with overlapping phenotypes with HCM and DCM [45]. It is most commonly attributed to mutations in seven genes(TAZ,DTNA,LDB3,LMNA,SCN5A,MYH7, andMYBPC3). With additional contributing variants reported in rare instances (ACTC1,TNNT2,MIB1,PRDM16, andTPM1), all LVNC loci encode proteins involved in cellular energy, muscle development, and ion channel formation, or are components of the muscle filaments. Despite these advances,many of the causative LVNC genes have yet to be identified. Mutations inMYH7are the single most common cause of LVNC, accounting for 8 to 13%of cases, with the remainder of the genes reported to be mutated in rare cases.
Genetic Testing of Inherited Cardiomyopathies
With the rapid development in genetic testing technology, an entire exome or a panel of cardiomyopathy related genes can now be tested simultaneously by the NGS, providing an opportunity to detect numerous mutations in same or different genes that are responsible for inherited cardiomyopathies. Several groups have developed NGS-based approaches for a comprehensive and cost-effective genetic diagnosis of cardiomyopathies. These approaches demonstrate the feasibility of using NGS techniques in targeted sequencing of cardiomyopathy associated genes [46, 47]. Multi-gene testing using NGS is a highly accurate and reproducible approach to the routine molecular genetic testing of patients with cardiomyopathies. The method has led to dramatic improvement in efficiency and speed of gene sequencing, as well as markedly reduced costs for clinical genetic testing [48]. Since th e application of NGS, the clinical genetic mutation detection rate has reached 32–47% for patients with HCM and 17–40% for patients with DCM[49–51]. However, one of the main challenges of NGS technologies is the analysis of the data.There is still variability concerning sensitivity and specificity within NGS platforms and different software, and Sanger sequencing still continues to be the gold standard for validation in ‘clinical sequencing’ experiments. With post-NGS Sanger sequencing confirmation, the analytic sensitivity and specificity of NGS in most CAP (College of American Pathologists) certified laboratory can reach 99.9 and 100%, respectively.
Genetic Testing of Hypertrophic Cardiomyopathy
Currently, genetic testing has been recommended for any patient with an established clinical diagnosis of HCM and for family members and appropriate relatives after the identification of the HCM-causative mutation in an index case [52].Including genetic testing in the diagnostic strategy is more likely to be cost-effective than are clinical tests alone when considering family screening and consequences for the prevention of SCD [53–55].Genetic testing identifies mutation carriers who will benefit from regular clinical investigations or early discussion of implantable cardioverter-defi-brillator (ICD), as well as relatives who do not have the causal mutation and, therefore, can be released without long-term follow-up [56]. With the rapid development in genetic testing technology, an entire exome or a panel of HCM-related genes can now be tested simultaneously by the NGS, providing an opportunity to detect numerous mutations in same or different genes that are responsible for HCM. Several groups have developed NGS-based approaches for a comprehensive and costeffective genetic diagnosis of cardiomyopathies and have been commercialized. For example, NGS pane ls testing services offered by Baylor College of Medicine (BCM)/John Welsh Cardiovascular Diagnostic Laboratory are listed in Table 2. Other laboratories provide similar tests including Ambry Genetics, GeneDx, etc. These approaches demonstrate the feasibility of using NGS techniques in targeted sequencing of cardiomyopathy associated genes [46, 47].
Genetic Testing of Dilated Cardiomyopathy
Because of significant locus and allelic heterogeneity, genetic testing for DCM has been of limited utility, with pathogenic variants identified in 17–40% of cases using current 40 gene panels, and most genes contributing only a small percentage of pathogenic variants [5]. Clear genotype-phenotype correlations are rare. Exceptions include variants in theLMNAandSCN5Agenes, which typically are associated with DCM and conduction systemdisease [57, 58]. With the application of NGS techniques,TTN-targeted sequencing revealed thatTTNtruncating mutations are a common cause of DCM [22]. Whole exome sequencing (WES) identified novel GATA Zinc Finger Domain Containing 1 (GATAD1) andBAG3mutations in patients with DCM [20, 59]. Due to the high level of complexity of genotype-phenotype associations and advanced genetic testing efficiency in DCM, genetic testing by NGS may be useful for the identification of noncarriers and asymptomatic carriers, as well as for prevention strategies, sport recommendations, and defibrillator implantation. It can also guide reproductive decision-making including utilization of pre-implantation genetic diagnostic strategies [60].Table 2 summarized the NGS panel for dilated cardiomyopathy developed in Baylor College of Medicine (BCM)/John Welsh Cardiovascular Diagnostic Laboratory.
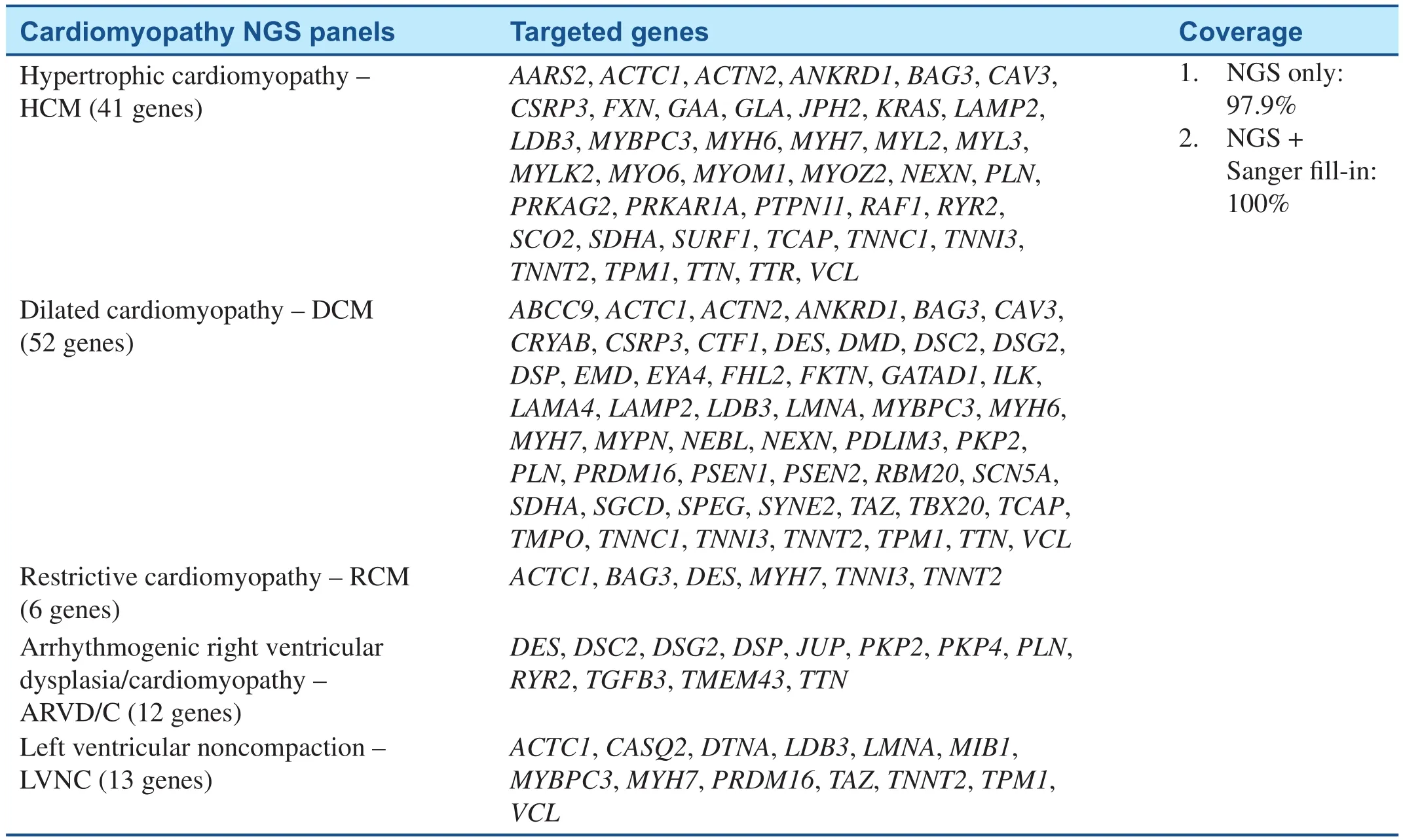
Table 2 Cardiomyopathy NGS Testing Panels in BCM/John Welsh Cardiovascular Diagnostic Laboratory.
Genetic Testing of RCM, ARVD/C,and LVNC
On the basis of previous studies, cardiomyopathy gene panels have been developed whereby a selected number of genes can be targeted and sequenced on NGS platforms commercially and are available for patients on demand (Table 2) [61]. The massively parallel approach of NGS technology allows the simultaneous sequencing of many genes; it is,therefore, an ideal technology for molecular genetic diagnosis of inherited cardiomyopathies and other genetically heterogeneous disorders. Additional Sanger sequencing is performed for any regions not captured or with insufficient number of sequence reads. All pathogenic and undocumented variants are con firmed by Sanger sequencing. Genetic testing may be useful to confirm the diagnosis of familial RCM [62]. Combining genetic testing with clinical screening of family members can greatly enhance the detection rate of familial LVNC to 67%[63]. LVNC genetic testing has no prognostic and therapeutic implications at this time, as clear genotype–phenotype correlations have not been well established [62].
Conclusion
Inherited cardiomyopathies are a group of inherited cardiac disorders with more than 80 genes identified to be involved so far. Although we noticed that there is genetic difference regarding the race,geography and ethnicity in inherited cardiomyopathies. This review discussed the disease-causing genes for inherited cardiomyopathy in general. Due to the limitation of pages and word counts and the limited data published so far in this regard, we did not expand to the genetic difference in population with different ethnicities. Genetic testing has long been used in clinical cardiology and a large number of genes and gene mutations have been defined to be related to specific inherited cardiomyopathies.NGS is perhaps one of the most exciting advances in the field of life sciences and biomedical research in the last decade. With the availability of massive parallel sequencing, a human DNA blueprint can be decoded to explore the hidden information. The new approach using NGS strategy allows a rapid molecular diagnosis for patients with cardiomyopathies at a broad coverage of the all known cardiomyopathy genes at a reasonable cost. Meanwhile,NGS is also a very powerful tool to be used to discover new genes related to cardiomyopathies through WES or WGS (whole genome sequencing).It is foreseeable that NGS technologies will be used increasingly in a clinical setting in the future. With more patients being tested by NGS, more and more cardiomyopathy-associated genes will be identified and this effort will further benefit our understanding of inherited cardiomyopathies and their clinical treatment.
Conflict of Interest
The authors declare no conflict of interest.
Funding Source
This study was sponsored by the internal research funding from Section of Cardiology, Department of Pediatrics.
REFERENCES
1. Maron BJ. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific statement from the Council on Clinical Cardiology,Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Function.Circulation 2006;113:1807–16.
2. Geisterfer-Lowrance AA, Kass S,Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990;62:999–1006.
3. Ho CY, Charron P, Richard P,Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res 2015;105:397–408.
4. Mcnally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest 2013;123:19–26.
5. Towbin JA. Inherited cardiomyopathies. Circ J 2014;78:2347–56.
6. Ware SM, Jefferies JL. New genetic insights into congenital heart disease. J Clin Exp Cardiolog 2012;Suppl 8:003. doi:10.4172/2155-9880.S8-003.
7. Maron BJ, Gardin JM, Flack JM,Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults : echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 1995;92:785–9.
8. Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH.Inherited cardiomyopathies. J Mol Diagn 2013;15:158–70.
9. Profiles P, Roberts WC, Mueller FO. Sudden death in young competitive athletes. J Am Med Assoc 1996;276:199–204.
10. Maron BJ, Doerer JJ, Haas TS,Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980–2006.Circulation 2009;119:1085–92.
11. Tester DJ, Ackerman MJ. Genetic testing for potentially lethal,highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation 2012;123:1021–37.
12. Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012;60:705–15.
13. Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy.Circulation 2010;122:2430–40.
14. Lopes LR, Zekavati A, Syrris P,Hubank M, Giambartolomei C,Dalageorgou C, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by highthroughput sequencing. J Med Genet 2013;50:228–39.
15. Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsuf fi -ciency. Circ Res 2009;105:219–22.
16. Towbin J, Lowe AM, Colan SD,Sleeper L, Orav EJ, Clunie S,et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. J Am Med Assoc 2006;296:1867–76.
17. Hershberger RE, Hedges DJ,Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–47.
18. Deck GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med 1994;331:1564–75.
19. Finsterer J, Stöllberger C, Wahbi K. Cardiomyopathy in neurological disorders. Cardiovasc Pathol 2013;22:389–400.
20. Norton N, Li D, Rieder MJ,Siegfried JD, Rampersaud E,Züchner S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet 2011;88:273–82.
21. Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD,Hofmeyer M, et al. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci 2010;3:90–7.
22. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P,Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–28.
23. Watkins H, Ashrafian H, Redwood,C. Inherited cardiomyopathies. N Engl J Med 2011;364:1643–56.
24. Morita H, Seidman J, Seidman CE.Genetic causes of human heart failure. J Clin Invest 2005;115:518–26.
25. Arbustini E, Diegoli M, Fasani R,Grasso M, Morbini P, Banchieri N,et al. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol 1998;153:1501–10.
26. DeWitt MM, MacLeod HM,Soliven B, McNally EM.Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J Am Coll Cardiol 2006;48:1396–8.
27. Mogensen J, Arbustini E.Restrictive cardiomyopathy. Curr Opin Cardiol 2009;24:214–20.
28. Chen S, Balfour IC, Jureidini S.Clini cal spectrum of restrictive cardiomyopathy in children. J Heart Lung Transplant 2001;20:90–2.
29. Russo LM, Webber SA. Idiopathic restrictive cardiomyopathy in children. Heart 2005;91:1199–202.
30. Rivenes SM, Kearney DL, Smith EOB, Towbin JA, Den field SW.Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation 2015;876–83.
31. Kaski JP, Syrris P, Burch M, Tome MT, Fenton M, Christiansen M,et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008;94:1478–84.
32. Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 2003;111:209–16.33. Chen Y, Yang S, Li J, Wang G, Qin Y, Wang D, et al. Pediatric restrictive cardiomyopathy due to a heterozygous mutation of the TNNI3 gene. J Biomed Res 2014;28:59–63.34. Peddy SB, Vricella LA, Crosson JE,Oswald GL, Cohn RD, Cameron DE, et al. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene.Pediatrics 2006;117:1830–3.35. Sm W, Me Q, Et B, Miller E, Uzark K, Rl S. Pediatric restrictive cardiomyopathy associated with a mutation in β-myosin heavy chain. Clin Genet 2008;73:165–70.36. Lopes LR, Elliott PM. Genetics of heart failure. BBA Mol Basis Dis 2013;1832:2451–61.37. Calkins H. Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ J 2015;79:901–13.38. Marcus FI, Edson S, Towbin JA.Genetics of arrhythmogenic right ventricular cardiomyopathy a practical guide for physicians. J Am Coll Cardiol 2013;61:1945–8.39. Campuzano O, Alcalde M, Allegue C, Iglesias A, García-Pavía P, Partemi S, et al. Genetics of arrhythmogenic right ventricular cardiomyopathy. J Med Genet 2013;50:280–9.40. Egle I, Li A, Bauce B, Nava A, Fanciulli M, Vazza G, et al.Identification of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy.Eur Hum Genet 2013;21:1226–31.41. Shemisa K, Li J, Tam M, Barcena J. Left ventricular noncompaction cardiomyopathy. Cardiovasc Diagn Ther 2013;3:170–5.42. Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium a study of eight cases.Circulation 1990;507–14.43. Bahler RC. Prevalence and characteristics of left ventricular noncompaction in a Community Hospital cohort of patients with systolic dysfunction. Echocardiogr J Cardiovasc Ultrasound Allied Tech 2008;25:8–12.
44. Ichida F. Left ventricular noncompaction. Circ J 2009;73:19–26.
45. Stress O, With I, Matter W, After R. HHS public access. Stroke 2014;44:3516–21.
46. Meder B, Haas J, Keller A, Heid C, Just S, Borries A, et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet 2011;4:110–22.
47. Dames S, Durtschi J, Geiersbach K, Stephens J, Voelkerding KV.Comparison of the Illumina genome analyzer and Roche 454 GS FLX for resequencing of hypertrophic cardiomyopathy-associated genes.J Biomol Tech 2010;21:73–80.
48. Waldmüller S, Schroeder C, Sturm M, Scheffold T, Imbrich K, Junker S, et al. Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies.Mol Cell Probes 2015;29:308–14.
49. Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB,et al. Results of clinical genetic testing of 2912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 2015;17:880–8.
50. Chalovich JM, Eisenberg E. NIH public access. Biophys Chem 2005;257:2432–7.
51. Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, et al. Genetic testing for dilated cardiomyopathy in clinical practice. J Card Fail 2012;18:296–303.
52. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H,et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society(HRS) and the European Heart Rhythm Association (EHRA).Heart Rhythm 2011;8:1308–39.
53. Wordsworth S, Leal J, Blair E,Legood R, Thomson K, Seller A, et al. DNA testing for hypertrophic cardiomyopathy: a costeffectiveness model. Eur Heart J 2010;31:926–35.
54. Ingles J, McGaughran J, Scuffham P, Atherton J, Semsarian C. A cost-effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart 2012;98:625–30.
55. Zhang L, Mmagu O, Liu L, Li D, Fan Y, Baranchuk A, et al.Hypertrophic cardiomyopathy: Can the noninvasive diagnostic testing identify high risk patients? World J Cardiol 2014;6:764–70.
56. Charron P. Genetic analysis for predictive screening in hypertrophic cardiomyopathy. Heart 2012;98:603–4.
57. Van Rijsingen I, Arbustini E, Elliott PM, Mogensen J, Hermans-Van Ast JF, Van Der Kooi AJ, et al. Risk factors for malignant ventricular arrhythmias in Lamin A/C mutation carriers: a European cohort study. J Am Coll Cardiol 2012;59:493–500.
58. McNair WP, Sinagra G, Taylor MR,Di Lenarda A, Ferguson D, Salcedo EE, et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism.J Am Coll Cardiol Elsevier Inc 2011;57:2160–8.
59. Theis JL, Sharpe KM, Matsumoto ME, Chai HS, Nair AA, Theis JD,et al. Homozygosity mapping and exome sequencing reveal GATAD1 mutation in autosomal recessive dilated cardiomyopathy. Circ Cardiovasc Genet 2011;4:585–94.
60. Mestroni L, Taylor MR. Genetics and genetic testing of dilated cardiomyopathy: a new perspective.Discov Med 2013;15:43–9.
61. Biswas A, Sandeep VR. Next generation sequencing in cardiomyopathy : towards personalized genomics and medicine. Mol Biol Rep 2014;41:4881–8.
62. Teo LY, Moran RT, Tang WH.Evolving approaches to genetic evaluation of specific cardiomyopathies. Curr Heart Fail Rep 2015;12:339–49.
63. Hoedemaekers YM, Caliskan K,Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, et al.The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet 2010;3:232–9.
杂志排行

Cardiovascular Innovations and Applications的其它文章
- The Role of Echocardiography in Hypertrophic Cardiomyopathy
- Rationale and Design of the Randomized Controlled Trial of Intensive Versus Usual ECG Screening for Atrial Fibrillation in Elderly Chinese by an Automated ECG System in Community Health Centers in Shanghai(AF-CATCH)
- Clinical Utility of Amlodipine/Valsartan Fixed-Dose Combination in the Management of Hypertension in Chinese Patients
- The Effect of Home-Based Cardiac Rehabilitation on Functional Capacity,Behavior, and Risk Factors in Patients with Acute Coronary Syndrome in China
- Depression, Anxiety, and Cardiovascular Disease in Chinese: A Review for a Bigger Picture
- Catheter Ablation of Atrial Fibrillation:Where Are We?