酶催化过程的全程模拟
2017-05-12曹泽星
赵 媛 曹泽星
酶催化过程的全程模拟
赵 媛1曹泽星2,*
(1河南大学天然药物与免疫工程重点实验室,河南开封475004;
2厦门大学化学化工学院,福建省理论与计算化学重点实验室,福建厦门361005)
酶催化包括底物到活性区的输运、选择催化化学反应及产物释放等复杂过程,由于复杂的蛋白质环境效应,任一化学和非化学过程都有可能是决定酶活性的关键步骤。为了全面认识酶催化活性,我们对几类酶催化过程进行了广泛的组合量子/分子力学(QM/MM)和经典分子力学(MM)动力学模拟(MD)研究,详细地讨论了整个酶催化过程的分子机制、关键残基的作用和蛋白质环境效应,丰富了对酶催化活性的认识。随着多尺度模型和计算模拟方法的进一步完善与发展,有望实现超大复杂生物酶催化过程的全程模拟研究,为酶工程领域的相关研究提供支持。
酶催化;底物输运;自由能计算;QM/MM MD模拟;随机加速动力学模拟
1 引言
1.1 酶催化特点及其调控因素
酶是一种具有生物催化功能的复杂高分子体系,是生物体中不可缺少的催化剂。如果酶缺失或酶活性降低,生命活动将发生紊乱,甚至无法维持1-9。通常酶催化具有很高的活性,其催化过程具有高度的专一性,即一种酶仅催化一种或一类底物参与的生物化学过程,并生成特定的产物。酶催化可以在温和的条件下进行,其活性与环境温度和pH值密切相关,温度和pH偏高或者偏低,都会显著影响酶的催化活性。如果遇到过酸、过碱或温度升高,酶的空间结构遭到破坏,就会导致酶变性失活。此外,催化活性还会受到多种其它因素调控,如抑制剂和激活剂调节、反馈抑制调节、变构调节和共价修饰调节等,有些酶的催化活性还与辅酶因子有关10。
近年来的研究发现,保守残基对底物结合和催化活性调控的作用机制、蛋白质构象的pH依赖性、底物和残基质子化状态等对酶催化效率的影响也是不可忽视的11-18。如6-磷酸葡糖胺脱氨酶(glucosamine 6-phosphate deaminase,NagB)中的催化三联体(catalytic triad)Asn128-His130-Glu135以及羟氰裂解酶(hydroxynitrile lyase,HNL)中的三联体Ser80-His235-Asp207,在催化过程中起着十分重要的作用,其酶催化最初的质子转移就是由这些保守残基的协同作用引发的16-18。HbHNL酶中的Thr11、Ile12、Ser80、Cys81、Leu157、His235和Lys236等残基通过与底物相互作用,将其限域在酶催化活性位点16;HbHNL和MeHNL酶中的残基Trp128在反应物进入活性位点及产物释放中,起到门控“开-关”的作用16,17;活性区赖氨酸质子化状态可以改变整个酶催化反应机制16,17;NagB中底物氨基的质子化状态将显著影响其催化N-乙酰葡糖胺(N-acetylglucosamine,GlcNAc)开环的机理,且反应活性口袋侧链残基His145的质子化状态会影响其lid motif区的“开-关”动力学行为,进而控制底物的结合,改变酶的活性18。
1.2 底物/产物输运与酶催化
在蛋白质环境中,酶催化全过程通常由一系列的化学步和非化学步组成,其中非化学步主要包括底物结合到活性位点的输运和产物从反应区域的释放(见图1)1-7,9,19。由于复杂的蛋白质环境效应,其中任何一个步骤都可能对酶催化效率产生重要影响。理论上,通过对酶催化过程的全程模拟,可以构造酶催化过程相对自由能变化的全景图,准确预测催化过程的决速步骤,深入了解酶催化微观机理,全面认识蛋白质环境中酶催化特性,进而有针对性地调控酶催化剂的活性。这将有利于仿酶催化体系的设计和相关的应用研究,在酶工程领域的研究中具有重要意义。
底物到活性区域的输运是酶催化过程的第一步,对酶催化过程有着不可忽视的影响,甚至有可能成为整个酶催化效率的决定性因素,近年来备受关注16,17,20-28。实验上主要通过晶体结构和残基突变分析等方法,推测可能的输运通道及关键残基,并结合瞬态动力学等方法预测底物传递的kcat值。比如在羟氰裂解酶(HNL)的研究中,实验研究组就通过晶体结构分析,预测了可能的底物传递通道,并通过将通道关键残基突变的方法,对其进一步验证29,30;在胞嘧啶脱氨酶的研究中,Yao等28通过瞬态动力学研究,推测产物释放可能是酶催化过程的决速步骤。然而,尽管如此,在实验上仍然很难获取底物或产物进出酶活性位点时的结构和酶构象的变化信息。理论上,随着分子动力学理论方法的发展,不但可以找到最为可能的反应通道,而且可以获取底物传递过程中的热动力学性质、关键残基和蛋白质构象变化,为实验研究提供理论支持20-27。最近,我们通过随机加速分子动力学(random acceleration molecular dynamics,RAMD)和传统分子动力学(classical molecular dynamics,MD)模拟,获得了HNLs酶催化过程中底物/产物输运的可能通道及传递过程中蛋白质的构象变化等信息16,17。

赵媛,1987年生。2015年于厦门大学获取理学博士学位,现为河南大学天然药物与免疫工程重点实验室讲师。现主要从事生物体系多尺度计算模拟及计算机辅助药物设计相关领域的研究。

曹泽星,1962年生。现为厦门大学化学化工学院教授,博士生导师。研究兴趣包括:激发态与光化学、无机与金属有机化学反应机理、复杂体系与酶催化过程的多尺度模拟、低维纳米材料的计算设计与模拟。

图1 酶催化过程(底物结合、化学反应、产物释放)Fig.1 Enzymatic process(substrate binding,chemical reaction,and product release)
酶催化反应机理是实验和理论都非常关注的焦点。实验上,主要采用X射线晶体衍射和核磁共振等实验手段,获取生物酶体系的结构,并通过同位素示踪、残基突变、酶反应动力学参数测定等方法表征酶催化反应机理。但是,由于很难捕捉催化过程中的过渡态和瞬时中间体的结构信息,加之酶静态晶体结构和活性态的结构差异,导致催化机制推测上的多样性及不准确性。随着理论与计算化学的发展,尤其是量子-经典力学组合方法(combined quantum mechanics/molecular mechanics,QM/MM)的不断完善,使得从原子和电子水平上解释酶催化反应机理成为可能,弥补了实验方法的局限性31,32。比如来自橡树和木薯的羟氰裂解酶(HbHNL&MeHNL)在碳碳键生物逆合成方面具有重要作用,二者高度同源,但是实验基于获取的原生态和突变体的晶体结构,推测两个酶具有不同的催化机制,且活性位点赖氨酸质子化对它们的调控作用也不一致33。而在理论上,基于酶相关的晶体结构,采用MM MD和QM/MM MD方法,建立了这两类酶活性态的初始结构,系统地研究了赖氨酸质子化状态对两类酶催化活性的影响,对实验推测的酶催化机理进行了一定修正,使之更能合理地解释有关实验现象16,17。
2 计算方法
2.1 MM MD与RAMD方法
分子动力学(molecular mechanics molecular dynamics,MM MD)方法主要基于牛顿经典力学建立多粒子系统的运动方程组,通过数值求解,模拟系统随时间推进的微观过程,获得系统粒子相轨迹,进而研究该系统的平衡热力学性质及结构动力学性质等。在MM MD模拟中,首先要搭建初始模型,并对其进行优化,消除初始模型中的不合理构象;然后对模型进行长时间分子动力学模拟,使系统达到平衡,并收集平衡后的数据,通过统计方法对系统的热力学性质等进行分析。其中,合理选取系统边界和粒子间作用势能模型、设定粒子初态、建立模拟算法计算粒子间作用力和各粒子的速度和位置等都有助于改进分子动力学模拟。常用的分子动力学模拟软件有Amber34、Namd35、Gromacs36-39、Charmm40、Tinker41和Lammps42等。
随机加速分子动力学方法核心在于对体系中处于活性位点的底物或产物分子质心或某个自定义原子上额外施加一个随机方向的力,推动其移动,直至离开整个酶体系,使其暴露于外部溶剂中,以此寻找底物或产物传递通道。在RAMD模拟中,给定初始的加速度及距离阈值,在一段时间内,当底物或产物达到或超过设定阈值,方向继续保持;否则,将施加另一个随机方向的力,以此来获取底物或产物分子传递的可能通道。但是,当随机作用力大于蛋白质系统对底物或产物约束力时,就有可能推动其朝向错误的方向,此时,就需要RAMD结合MD的方法来解决这个问题。当底物或产物受力逃离初始位置,经典MD模拟就会开启平衡整个体系,在一定程度上纠正过强的随机作用力,使底物朝正确方向移动。在RAMD模拟中,加速度和阈值的设定非常重要,为了获得合理可行的模拟结果,通常需要设置多个加速度、多个阈值,根据轨迹统计,来获取通道情况。目前可用Amber34、Namd35、Gromacs36-39和Charmm40来实现RAMD的计算。
目前,将MM MD和RAMD相结合是研究底物输运可能通道及其相应热动力学性质的重要方法之一,在酶催化过程的全程模拟研究中具有重要意义15-17。
2.2 QM/MM与QM/MM MD方法
近年来,量子-经典力学(quantum mechanics/ molecular mechanics,QM/MM)方法已经成为研究酶催化过程中化学反应步的强大工具43-60。其中,QM方法用来描述关键区域(quantum mechanics subsystem,QS),如化学反应活性区域,MM方法用来描述活性区域环境(molecular mechanics subsystem,MS),如周围蛋白质环境及溶剂。QM/ MM方法可以在考虑蛋白质和介质环境的前提下,从原子及电子水平上合理描述酶催化体系的结构、反应机理及其能量学性质(图2)。而QM/MM MD则是在QM/MM的水平上进行分子动力学模拟,并结合伞形采样(umbrella sampling,US)等技术,可以获得蛋白质和介质环境动力学对催化反应过程的影响及其反应过程的自由能性质,相对于QM/MM计算获得的静态结构和能量信息而言,自由能变化可以更加合理地描述酶催化反应的特征。值得注意的是,当体系划分为QS区和MS区之后,二者之间会形成边界,如何描述两个区域间的边界相互作用也是QM/MM方法一直关注的问题。
QM/MM方法计算体系的能量时,通常采用减法方案和加法方案。减法方案的能量表达式为:
EQM/MM=EMM(MS+QS)+EQM(QS)-EMM(QS)其中,EMM(MS+QS)为MM水平下获得的MS和QS区域的总能量,EQM(QS)为QM水平下QS区域的能量,EMM(QS)为MM水平下QS区域的能量。它的优点是在QS区和MS区之间没有耦合项,处理较为简单直接。缺点是在QS区也采用力场来处理,在一些情况中不可行。而且当有化学反应发生时,QS区电荷分布会发生变化,在处理静电相互作用中,用固定原子点电荷描述是不准确的。此外,无法考虑MS区对QS区电子密度的极化。Morokuma等61,62提出的ONIOM方法运用此种方案。加法方案的能量表达式为:

图2 QM/MM系统Fig.2 General QM/MM system
EQM/MM=EQM(QS)+EMM(MS)+EQM/MM(QS/MS)其中,EQM(QS)和EMM(MS)为QS区域和MS区域能量项,EQM/MM(QS/MS)为QS和MS区域相互作用能,一般包括成键相互作用项和非键相互作用项,而非键相互作用项又包括静电相互作用和范德华相互作用。静电相互作用是处理耦合的关键,通常采用机械嵌入(mechanical embedding)、静电嵌入(electrostatic embedding)和极化嵌入(polarized embedding)来处理。在大多数机械嵌入中,QS区域是孤立的,其电子密度不会受到MS区域的影响,且处理QS及MS之间相互作用是在MM水平下进行的。其优势是减小了计算量,但较为粗糙。ONIOM(MO:MM)方法61,62则通过这种嵌入方案来处理QS和MS区域间的静电耦合。静电嵌入方案则是机械嵌入方案的改进,在此方案中,将QS与MS之间静电相互作用当做单电子算符考虑到QM哈密顿算符中。此时,QS区电子结构会被MS区的电荷分布所极化,提高了计算精度。在极化嵌入方案中,两个区域可以相互极化,即MS区也会被QS区域所极化。常用的模型有charge-on-aspring model63,induced dipole model64和fluctuating charge model65。在这种方案下,要获取总能量,在QM波函数每一步自洽场迭代都需要进行一次MM极化计算。这种处理非常耗时,也会带来收敛问题。因此,此方案往往适用于MS区域较小的体系66。
针对边界处理,目前较为流行的处理方法有:连接原子法(link-atom scheme)67-72,边界原子法(boundary-atom scheme)60,73-78和定域轨道法(localizedorbital scheme)43,46,53,79-84(图3)。连接原子法是处理QM与MM边界问题最直接的方法。它在QM与MM切断处Q1―M1引入一个额外的原子中心或一个悬空的键B来满足Q1的自由价态。通常采用氢原子,但也可以是任何单价原子或基团。其中,电子饱和部分包括B和Q,采用QM处理;Q1―M1键用MM描述。边界原子法是利用一个特殊的类似“两面神”的边界原子Cps来代替M1原子,既作为正常的MM原子参与MM计算,又用来饱和Q1的自由价态,出现在QM计算中。它可以避免连接原子法中引入额外原子的问题,还能够模仿边界上MM基团的电子特征。大多数边界原子法的提出都基于单价赝势(或有效势),赝势局域在M1原子的位置,可利用参数化来重现特定期望的性质,如利用C―C单键的截断来模仿甲基。目前,这类方法也有许多发展,如调节原子(adjusted connection atom)75、赝键(pseudobond)60,73,74,85、有效团势(effective group potentials)76,86-88、量子覆盖势(quantum capping potentials)78,89、极小原理有效哈密顿(effective hamiltontians from a minimum principle)77、多中心价电子有效势(multicentred valence-electron effective potentials)90等。定域轨道法是采用一个冻结的杂化轨道来饱和QM-MM边界的悬空键。其共同点是在一个前线原子上设置一组合适的定域轨道,并保持这些轨道冻结,不参与SCF迭代。这类方法主要有局域自洽场(local self-consistent field)80-82,84,91、冻结轨道(frozen orbitals)53,54,92、广义杂化轨道(generalized hybrid orbitals)46,83,93-95和有效碎片势(ffective fragment potentials)79,96-98等。

图3 (a)连接原子法;(b)边界原子法;(c)定域轨道法Fig.3 (a)Link-atom scheme;(b)boundary-atom scheme; (c)localized-orbital scheme
2.3 自由能计算方法
自由能与实验数据有直接可比性,是构造酶催化过程全景图不可缺少的数据之一,通常可分为亥姆霍兹自由能(A)或吉布斯自由能(G)。亥姆霍兹自由能适合正则系综(canonical ensemble,即NVT系综),吉布斯自由能适合NPT系综,其中,ΔG=ΔA+ΔPV。生物大分子是一个凝聚相体系,因此,可将ΔPV近似为0,那么ΔG≈ΔA。对于蛋白质环境中的化学反应,自由能不能只基于静态电子结构计算获取相对能量值,而需考虑到体系的涨落,合理描述反应的动态过程。
目前,自由能计算方法种类较多,如伞形采样99-102、自由能微扰103-105、热力学积分(thermodynamic integration,TDI)106、metadynamics107-109和Jarzynski equality110-113等。US主要通过施加偏势来改变势函数,使得体系能够在高能区采样,而后根据概率密度求解自由能数据,较多应用于对新提出的自由能计算方法准确性检验111,112,114-116;FEP基本思想是在初末态间插入若干中间态,通过获取各态间自由能变化,来获取初末态自由能变化,这种方法原理严格,不需要规定反应路径,对于小分子来说较为精确117-119;TDI与FEP类似,只是对两个状态间自由能差的定义不同,利用各态能量系综平均值获得自由能,近年来,自适应偏置力(adaptive biasing force,ABF)方法120-122就是在TDI的基础上发展起来的,它引入了偏置力概念,提高采样效率,且采样均匀;metadynamics方法主要通过对体系低自由能区不断施加额外的高斯型排斥势能函数,使其到达高自由能区域,进而通过施加的势能函数来对自由能进行计算,此方法不需要事先对反应路径有预先判断,因此,在寻找最优反应路径方面有较大优势123;Jarzynski equality方法是需要施加额外的恒速运动谐振势使得体系到达高自由能区进行采样,由大量的不可逆功的系统平均来求解反应过程的自由能,在操作便捷及计算效率上有较大优势111。上述几种方法在自由能势能面构造中,均得到了广泛应用111,112,114-124。
2.4 关键残基作用分析方法
关键残基在底物/产物输运及酶催化化学反应过程中起着十分重要的作用,通常采用残基突变方法,讨论保守残基在酶催化过程中所扮演的角色。例如,将重要残基突变为丙氨酸,通过QM/ MM计算或MM MD模拟,以及和原生酶体系结果的比较,可以探明关键残基在酶催化过程中不同阶段所起的结构或功能作用。
理论上,评估关键残基在底物结合中的作用,常采用molecular mechanics generalized born surface/ poisson-boltzmann surface area(MM-GB/PBSA)方法。该方法是基于分子力学与连续介质模型的一种结合自由能计算方法,已成功应用于蛋白质-配体125-130,蛋白质-蛋白质131-133,蛋白质-肽相互作用134-136的研究中。配体(L)和受体(R)形成复合物RL的结合自由能(ΔGbind)的计算如下:
ΔGbind=ΔH-TΔS≈ΔEMM+ΔGsol-TΔS
其中,ΔEMM、ΔGsol和-TΔS分别代表气相MM能量变化、溶剂自由能变化和结合熵变。ΔEMM包括键长、键角和二面角能量、静电相互作用能和范德华相互作用能。ΔGsol是溶剂化能总和,包括采用GB和PB模型计算得到的极化溶剂化能(极化部分)和采用溶剂可及表面积(solvent accessible surface area,SASA)得到的非静电溶剂化能(非极化部分)。
3 计算研究进展
3.1 羟氰裂解酶催化过程中自由能变化全景图的构造
羟氰裂解酶(hydroxynitrile lyases,HNLs)能够催化氰醇生成相应醛(酮)及氢氰酸(HCN),HCN的释放不仅可以帮助植物抵御食草动物及微生物的侵害137,而且能够为天冬氨酸的生物合成提供氮源138。此外,其逆反应可以用来合成手性化合物,并具有较高选择性139-142。近几年,HNLs已经成功应用于药物的生物逆催化合成143-149。因此,对HNLs催化过程的深入研究将有助于工业产物优化及生物催化设计。最近,我们采用MM和QM/ MM MD的方法,结合伞形采样技术,对来自木薯的羟氰裂解酶(hydroxynitrile lyases from Manihot esculent,MeHNL)催化过程进行了全程模拟,获得的自由能变化全景图见图4(a)17。整个酶催化过程包括底物氰醇到酶活性位点的输运、羟氰裂解成丙酮和氢氰酸和产物释放三个步骤。
对于底物/产物的输运等非化学步,通过RAMD MD模拟,探明了反应物进入活性位点和产物释放的可能通道及其重要性17。计算模拟中,定义中心碳原子为丙酮氰醇、丙酮和HCN的近似质心,采用 20.9、18.8、16.7、14.65、12.56、10.47和8.37 kJ·nm-1·g-1的加速度和0.0005 nm的阈值来驱动底物/产物的传递和可能通道确定。对于底物输运和产物释放,分别获得28条轨迹,并发现三条可能通道(分别定义为通道A、B、C)。通道A位于残基180-190和Trp128之间,通道B位于残基115-130和Trp128之间,通道C位于残基115-130和210-225之间,三个通道距离蛋白质边缘的距离分别为1.2、1.2和1.5 nm。而且,通道A的口袋比B和C宽,由此推测通道A是最具有优势的传递通道(见图4(b))。通过对底物的通过几率进行统计,进一步验证了通道A具有绝对的优势。在底物和产物的输运过程中,通道A的贡献在百分之八十以上,而落到通道B和通道C的轨迹非常少。在此基础上,通过MM MD方法结合伞形采样技术,对底物/产物通过主要通道输运的能量性质进行了计算分析,分别获得了一维和二维相对自由能图(见图4(c))及其详细的输运机制。
计算模拟揭示,反应物结合至活性位点依据Trp128的翻转可分为四步(见图4(b))。首先,反应物逐渐靠近蛋白质,并与Gln181和Trp128分别形成氢键和非键相互作用,推动底物进入蛋白质通道,此时,Trp128和Met147处于关闭的状态;然后,Trp128逐渐翻转,通道打开,底物和Trp128的共轭环形成lone pair…π和π…π相互作用,并与Met147形成静电相互作用;接下来,底物的―CH3基团和Trp128形成―CH3…π相互作用,Trp128逐渐关闭;最后,底物结合至活性位点,与Thr11、Ser80和 Lys237形成氢键相互作用,Ile12、Leu149、Leu158和Ile120形成疏水口袋来容纳反应物的―CH3基团,此时,Trp128完全关闭。
HCN和丙酮的释放可分为六个阶段。第一阶段,HCN离开活性位点,朝着通道开关移动,Trp128在“开-关”态之间转换;第二阶段,HCN穿过通道开关区域,Trp128逐渐关闭;第三阶段,HCN完全释放,体系能量到达局域最小值点;第四阶段,丙酮试图离开活性位点,并与Trp128形成―CH3…π、lone pair…π和π…π相互作用,驱使Trp128由闭态转换为开态;第五阶段,丙酮沿通道离出,Trp128逐渐恢复关闭状态;最后,丙酮完全释放,暴露于水环境中,通道完全关闭。
酶催化化学反应过程可分为三步(见图4(d)),首先是双质子转移,即质子由底物传递给Ser80,Ser80又将自身的质子传递给His236,形成两性离子中间体组态IM1,经过采样统计,明确了双质子转移是同时发生的;其次是C―CN键断裂,形成中间体IM2,此时,氰根阴离子(CN-)形成,并被Lys237的―NH3+基团稳定,形成强静电相互作用;最后是HCN的生成,即Pc态,也经历了双质子转移,即质子由His236传递至Ser80,随后Ser80将自身质子传递给CN-。
从构造的自由能变化全景图来看,反应物从酶外部进入活性位点,形成底物结合的B态,释放51.9 kJ·mol-1能量,是整个酶催化过程的驱动力之一;催化化学反应步的能垒主要来自于C―C键的断裂和HCN的生成,自由能跨度约为71.6 kJ· mol-1,决定了整个酶催化的进程;产物释放二维自由能图(见图4(c))揭示,HCN的释放先于丙酮,HCN释放能垒约为8.4 kJ·mol-1,且放热19.7 kJ· mol-1;丙酮释放能垒约为41.9 kJ·mol-1,在一定程度上影响了酶催化效率,当释放的产物分子进入到水介质环境,强的溶剂化作用将释放出显著的能量,有助于酶催化过程的进行。
3.2 核苷水解酶催化过程自由能变化全景图的构造
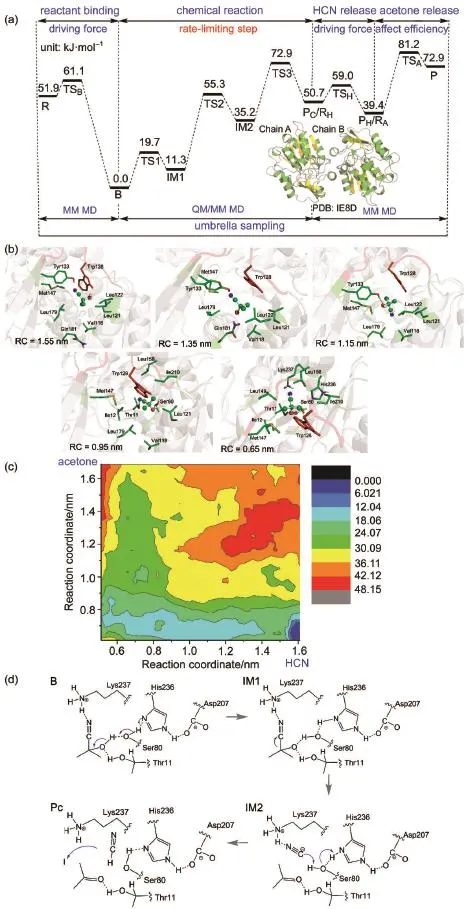
图4 (a)催化过程自由能变化全景图17;(b)底物输运过程中具有代表性态的关键残基及构象变化17;(c)产物释放自由能变化图17;(d)酶催化中的化学反应过程17Fig.4 (a)Free energy profile of the whole enzymatic catalysis17;(b)key residues and their conformational changes at the representative states in the main channel17;(c)free energy profile of product release17; (d)chemical reaction steps involved in enzymatic process17
核苷水解酶(nucleoside hydrolases,NHs)是一类含金属离子Ca2+的金属酶,在病原生物体嘌呤补救中扮演重要角色150,151,可以有效催化核糖核苷N-糖苷键水解生成核糖和碱基,因此,成为非常具有吸引力的抗寄生虫靶标152,153,其水解反应过渡态类似物常用来设计NHs抑制剂。肌苷-腺苷-鸟苷-核苷水解酶(IAG-NH)是NHs中的一类,倾向于催化肌苷(inosine)、腺苷(adenosine)和鸟苷(guanosine)的水解154。实验预测IAG-NH的催化过程可分为四个步骤,分别为底物结合至酶活性位点、化学反应、碱基释放和核糖释放155-157,其中核糖释放可能是整个过程的决速步骤155,158。近期,Wu等11,15,159对IAGNH的催化过程进行了详细研究,构造了其自由能全景图(见图5(a))。
QM/MM计算模拟表明11,肌苷在IAG-NH的催化作用下水解生成糖环和碱基,其中,糖环和碱基之间C―N键的断裂以及糖环和水分子之间的C―O键生成是同时发生的(见图5b),底物N7位的质子化可以显著地降低化学步的能垒,预测的自由能能垒约为29.3 kJ·mol-1;随后,碱基释放所需的能量约为23.4 kJ·mol-1,显然这些过程对整个酶催化过程影响不大159。最后,核糖的释放包括核糖脱离Ca2+的配位形成自由态、周边水分子替代核糖参与Ca2+配位和核糖穿过酶通道完全释放三个步骤15。其中,自由态核糖通过通道完全释放能垒约为54.4 kJ·mol-1,与估算的实验值15569.9 kJ·mol-1较为吻合,是整个催化过程的决速步骤。

图5 (a)催化过程自由能变化全景图11,15,159;(b)IAG-NH催化肌苷水解机制11;(c)产物传递过程具有代表性态的关键残基及构象变化15Fig.5 (a)Free energy profile of the whole enzymatic catalysis11,15,159;(b)hydrolytic mechanism of inosine catalyzed by IAG-NH11;(c)key residues and their conformational changes at the representative states for the product release15
为了深入了解糖基释放决速步骤的相关信息,应用RAMD MD方法对核糖释放通道进行了详细研究15,动力学模拟采用了20.9、12.6和8.4 kJ·nm-1·g-1的加速度及0.0002和0.0004 nm的阈值,获得18条轨迹。计算发现两条通道(见图5b),主要通道在loop 1和loop 2之间,有13条轨迹落在其中,比例为72%;次要通道在loop 2和残基17-184之间,轨迹比例为28%。值得注意的是酶活性位点距离主要通道和次要通道的距离分别为1.1和0.6 nm,为了探索距离长的反倒成为主要通道的原因,分别对它们进行了详细研究,发现主要通道以极性残基为主,如Asn12、Asp14、Glu82、His246和Arg252等,而次要通道则以非极性残基为主,如 Trp185、Trp260、Phe79和Phe175,后者位阻较大,而且从通道宽度而言,主要通道优于次要通道。此外,研究通过MM MD方法和伞形采样技术获得了糖环释放过程主要通道的热动力学性质,结果显示,随着Trp83的“开-关”振动,一些水分子逐渐进入通道,造成糖环与通道残基形成直接或间接的氢键相互作用,尤其是与Arg252之间的氢键相互作用导致其驻留时间增长;而且,通道主要残基侧链芳环较大,有较强的位阻效应。因此,二者共同阻碍了糖环的释放,是核糖释放能垒的主要来源。
3.3 底物及残基质子化对催化的影响

图6 SmuNagB催化GlcN6P开环自由能曲线Fig.6 Relative free energy profiles of the ring-opening reaction along the reaction coordinate for SmuNagB color online
6-磷酸葡糖胺脱氨酶(NagB)属于醛酮异构酶,能够催化6-磷酸葡糖胺(GlcN6P)转化为6-磷酸果糖(F6P)和氨分子,决定了N-乙酰胺葡萄糖(GlcNAc)的代谢方向160。实验上对其蛋白质结构、动力学性质、lid区柔韧度、变构调节、基因重组、功能分析和催化机理进行了广泛研究161-167,并发现其催化活性与pH值密切相关,在pH=7.5-9.5时,酶的催化速率达到最大(kcat>30 s-1),当pH〈6时,完全失去酶活性162。根据质子化葡糖胺的pKa值168,GlcN6P的氨基存在两种质子化态,分别为―NH2和―NH3+。因此,理论上,针对这两个态,分别建立去质子化态模型和质子化态模型,并采用QM/MM MD方法结合伞形采样技术对两个模型的GlcN6P的催化开环机理进行研究,获得了二者参与开环过程的自由能图(见图6)。QM/MM MD结果显示,对于去质子化态模型而言,GlcN6P将质子传递给His130与环C1―O5键断裂是同时发生的,能垒约为87.9 kJ·mol-1。对于质子化态的底物模型,开环为分步过程。首先,质子从底物的O1转移至His130的Nε,产生两性离子中间体(IM1),能垒为8.8 kJ·mol-1,此时,底物―NH3+能够稳定O1-。随后,六元环C1―O5键断裂,形成开环中间体IM2,能垒为75.4 kJ·mol-1,为开环反应的决速步骤。因此,质子化的底物GlcN6P比去质子化态的形式更易发生开环反应,且形成的中间体更稳定。开环产物分解释放氨并与介质水强的溶剂化作用,将显著地稳定产物态,促进酶催化开环过程。
对于来自橡树(HbHNL)和木薯(MeHNL)的羟氰裂解酶(hydroxynitrile lyases,HNLs),两类酶具有很高的同源性(77%sequence identity)33,然而,基于酶与复合物突变体系的晶体结构,实验上建议两类酶催化具有不同的反应机制。在HbHNL催化过程中,活性区域赖氨酸的质子化态被认为具有十分重要的作用,然而对于MeHNL催化,实验基于Ser80Ala突变体与氰醇复合物的晶体构型(PDB ID:1E8D),认为活性位点赖氨酸对催化并无作用33。为了探明两类酶催化机制的差异,基于MM MD模拟获取的wild type酶与底物复合物的稳定构象,采用QM/MM MD方法,并结合伞形采样技术,我们对HbHNL和MeHNL的催化反应机理进行了系统研究16,17。计算模拟中,通过指定活性位点关键残基赖氨酸不同的质子化状态,构建两类酶的质子化和去质子化计算模型,以调查赖氨酸在催化过程中扮演的角色。我们的研究表明,对两类酶催化过程,赖氨酸的质子化状态显著地影响酶催化微观机制。当酶活性区的赖氨酸处于质子化状态时,其化学步包含双质子传递(底物→Ser80→His236)、C―C键断裂和HCN形成三个步骤,其中,一个显著特点是在C―C断裂之后,氰根阴离子(CN-)生成,并与活性位点赖氨酸的形成强静电相互作用;对于去质子化态模型而言,首先经历了双质子传递,随后,C―C键断裂和HCN生成是同时发生的,并且由于赖氨酸的―NH2基团为中性,无法稳定CN-,因此,催化反应中并无CN-
生成。从反应能垒上看,HbHNL和MeHNL的质子化态模型的总能垒分别为75.4和71.2 kJ·mol-1,而去质子化态模型分别为92.1和129.8 kJ·mol-1。由此可见,活性位点赖氨酸的质子化态在两类酶催化反应中都具有重要作用。
上述实例讨论了底物或残基质子化状态的改变对酶催化过程的影响,很明显,关键基团质子化形式的改变,不但影响了整个酶催化效率,甚至完全改变了催化反应机制,这种现象是酶催化蛋白质环境效应之一,也可能出现在其他的酶催化体系中。
3.4 残基质子化态对酶关键区域构象动态行为的影响
近几年,来自大肠杆菌(Escherichia coli)和人类(Homo sapiens)的NagB六聚体及来自枯草芽孢杆菌(Bacillus subtilis)和变形链球菌(Streptococcus mutants)的NagB单体的晶体结构被相继确定,但在现有的晶体中,仅有一个为酶-底物复合物的形式(PDB ID:2RI1)162(来 自 Streptococcus mutants,SmuNagB),它是在酸性溶液中获得的。晶体结构揭示,在活性区域结合口袋开口边缘处存在一个灵活易变的lid motif,可以导致结合口袋处于“开态(open state)”或稳定的“闭态(close state)”161,169。一旦活性口袋处在闭态,底物和产物无法进出酶催化活性区域。实验上,SmuNagB酶催化D-葡糖胺-6-磷酸(GLcN6P)脱氨的活性与pH值密切相关,最高活性出现在pH=8.0-8.5的区间。为了理解lid motif调控活性口袋开关态的微观机制及酶催化活性与pH值关系的本质,我们对这类酶催化进行了广泛的计算模拟研究。
基于SmuNgaB酶的apo态及SmuNagB-GlcN6P酶-底物复合物的晶体结构(PDB ID:2RI0,2RI1)162,建立了两类计算模型18,分别代表酶的自由态和复合态,酸性条件下模型为Model A和Model B,碱性条件下的模型为Model C和Model D。为了减少计算量,仅考虑其中一条链。模拟研究发现,当体系平衡之后,底物进入酶的活性位点(Model B),lid motif处于结合口袋上方,形成close态;然而在其他三个模型中,活性区域口袋均为open态。残基结构涨落(RMSF)分析显示,对于Model B,lid motif(由残基152-168组成)的柔韧度非常小,而Model C和D柔韧度相似。进一步,活性区域结合口袋体积估算结果显示,在酸溶液中,当底物结合至活性位点时,lid motif经历了从open到close的动态变化(图7)。但是,Liu研究组162获得的酶-底物复合物晶体结构中的lid motif构型与apo态近似。通过比较MD模拟中不同阶段的构象变化及RMSF值,我们发现,在酸性条件下,随着模拟的进行,lid motif逐渐靠近底物存在的结合口袋,逐渐导致close态的形成,其柔韧度也逐渐降低,与EcoNagB及BsuNagB类似,都在底物结合时经历了类似的构象变化。和酸性溶液中不同的是,在碱性条件下,酶结合底物后,lid motif仍远离活性通道,结合口袋处于open态。
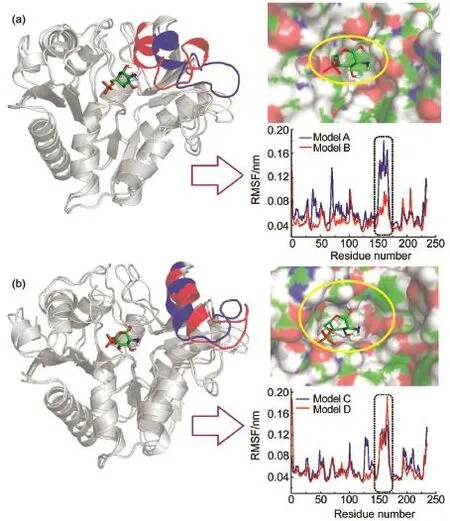
图7 酸性(a)和碱性(b)溶液中,SmuNagB的apo态(蓝)和复合态(红)的叠合图、口袋表面图和对应的RMSF值18Fig.7 Overlap for the apo state(blue)and the enzyme-substrate complex(red)in acidic(a)and basic solutions(b)along with RMSF values of the backbone and surface of the active site pocket18RMSF:root mean square fluctuation.color online
为了进一步理解lid motif动态行为的pH依赖性,理论上对活性口袋和lid motif之间的氢键网络进行了统计。如图8所示,在酸性溶液中,对复合态而言,Asn157和底物之间通过水分子形成了氢键网络,使lid motif向活性位点靠近。而且Thr150&Glu135,Asn134&Hip145形成氢键的概率分别为32%和99.7%。在采样过程中,四个残基对lid motif的关闭起到诱导作用。但在apo态中,并未见此现象,lid motif始终停留在open态。因此,酸性溶液中,lid motif的构象变化来源于底物的结合。在碱性溶液中,无论酶的复合态还是apo态,lid motif总是停留在open态。而lid motif和活性位点之间的区域并无氢键网络使lid区关闭,预测碱性和酸性溶液中的行为有所不同,可能在一定程度上受到了His145质子化状态的影响。His145双质子化使lid motif区与活性位点附近残基形成氢键网络,导致酸性溶液中的close状态。但是,在碱性溶液中,氢键相互作用消失,lid motif区为open态。因此,从理论上可以推测出lid motif在酸碱条件下的不同动力学行为主要来源于His145的质子化态及其导致活性位点氢键网络的重排。
3.5 关键残基在酶催化过程中的调控作用

图8 Model B(a),ModelA(b)和Model D(c)的活性位点及lid motif区氢键网络详情18Fig.8 Hydrogen bond networks around the active site and the lid region of Model B(a),ModelA(b),and Model D(c)18
实验研究表明,催化三联体(Asn128-His130-Glu135),在NagB酶催化GlcN6P的开环反应中扮演了重要角色,其中,His130是中性的,它在开环过程中,诱发质子转移,且Asn128及Glu135单突变均能完全改变酶反应活性162。为了合理评估Asn128及Glu135在催化反应中的作用,理论上分别对Asn128Ala和Glu135Ala的单突变及双突变体系进行了QM(B3LYP)/MM计算18。
对于双突变体而言,去质子化态双突变体系能垒比原生物酶体系高42.7 kJ·mol-1,质子化态双突变体系质子传递及C5―O1键断裂比原生物酶体系高18.0和167.1 kJ·mol-1。酶活性消失主要来自于突变造成的活性位点氢键及极化相互作用的改变。对质子化态模型而言,Asn128和Glu135突变将明显降低酶活性,这与实验观测一致,同时也意味着质子化态模型在催化过程中比重较大。在单突变体中,对去质子化模型而言,Asn128Ala突变体的能垒比原生体系高40.0 kJ·mol-1,而Glu135Ala突变体则与原生体系较为接近;对质子化模型而言,Asn128Ala和Glu135Ala单突变体系质子转移的能垒相对原生物酶体系分别增加25.5和12.6 kJ·mol-1,C5―O1键断裂的能垒则分别增加210.2和193.8 kJ·mol-1。两个结果都显示Asn128在催化过程中比Glu135起到更重要的作用。此外,电荷分析证明Glu135Ala突变体较Asn128Ala而言,更能加强C1―O5键的极化作用,在一定程度上解释了Glu135Ala单突变体键断裂能垒与Asn128Ala相比较低的现象。
对于HbHNL酶催化,实验上推测,如果将Trp128突变为Ala,将有利于更大的芳香氰醇进入活性位点170,171。理论上,通过MM MD模拟并结合伞形采样技术,也证实Trp128在底物输运中起到开-关的作用16。为了进一步探索Trp 128在底物结合及产物释放中的功能及其对酶催化效率的影响,预测了Trp128Ala突变体系中底物输运的自由能图,发现底物释放需要跨过33.5 kJ·mol-1的能垒,仅是原生酶体系(~67.0 kJ·mol-1)的一半。很明显,虽然其他疏水残基氢键网络在一定程度上影响了底物传递,但Trp128的吲哚环带来的位阻效应及其翻转仍是能垒的主要来源。
除此之外,关键残基在底物结合中同样起着十分重要的作用。例如,在HbHNL酶催化的反应机理研究中,Gruber等33指出,Lys236、Ser80和Thr11与底物存在强的氢键及静电相互作用,在底物结合中起着重要作用。为了进一步了解每个残基的影响,我们对K236A、S80A、T11A突变体系和原生HbHNL体系的底物结合自由能进行了估算与分析16。计算结果表明,范德华、静电相互作用和非极性溶剂效应为底物结合提供了主要驱动力;而极性溶剂化能和熵效应则起了负作用。强静电相互作用抵消了极性溶剂化能的作用,对C―CN键的活化及断裂而言非常重要。和原生HbHNL体系相比,K236A,S80A和T11A突变体系的结合自由能分别减小了12.4、19.8和5.1 kJ·mol-1,这可能主要来自于静电相互作用。此外,从per-residue模式对各个残基进行了自由能能量分解来看,位于活性位点的Ile12、Cys81、Leu157和His235对底物结合有显著作用,其中,范德华及非极化溶剂能为主要贡献。
4 结论与展望
本文简要总结了我们近年关于几类酶催化过程全程模拟研究的进展。通过构建合理的MM和QM/MM多尺度模型,基于经典和量子-经典动力学模拟,实现了对整个酶催化过程微观机理的理论描述。模拟研究不仅可以预测酶催化化学反应过程的机理,而且能够获得底物输运与产物释放过程的分子机制及其能量学性质,探明蛋白质构象变化和关键残基对酶催化的影响,为酶活性的调控、酶抑制剂的合理设计、仿酶催化新体系的发展等酶工程领域的相关研究提供理论依据。
基于全程模拟,可以获得底物输运通道的完整图像和关键残基的作用机制,有助于对酶和底物的结构进行针对性的改造,加强或减弱底物与通道侧链残基间的相互作用,实现底物选择性的传递。对生物逆合成有机分子而言,在关键残基的驱使下,使得具有不同官能团的有机分子进入酶活性位点,可以在一定程度上丰富分子合成的种类。在药物分子设计中,可以根据获得的残基和底物作用模式,设计具有特定官能团的药物分子,延长药物-靶标作用时间,药物-靶标作用时间的调控在药物结合动力学中具有重要意义,这也是实验研究较为关心的问题之一172。此外,基于对酶催化化学反应步模拟获得的过渡态电子结构及其与周围氨基酸残基结合模式等信息,可以设计一个过渡态类似物,使其与酶紧密结合,而设计出的分子极有可能成为该酶的抑制剂,这也是基于机制或结构的现代计算机辅助药物设计方法之一173-175。近期,Zhou等176通过QM/MM MD模拟,探明了HDAC酶催化反应的微观机理,并发展了一种基于反应机理设计抑制剂的策略,设计出HDAC2选择性抑制剂β-羟甲基查尔酮,而且,通过实验验证其结合能力较强,是HDAC1的20倍。
量子-经典力学(QM/MM)组合方法是目前研究酶催化反应最为流行的方法之一,但随着QM区域的增大,计算量也会显著增加,尤其是QM/MM MD模拟,很难采用大的QM分区。此外,在QM/ MM中,MM部分的原子电荷一般都是非极化的,无法正确地描述QM和MM区域的相互作用,影响QM/MM的计算结果。目前的QM/MM方法仍存在许多挑战,还需要发展理论新方法,合理描述QM/MM方法中多尺度边界和长程QM-MM Coulomb相互作用,实现低标度QM计算和多尺度的快速QM/MM MD模拟,提高QM/MM计算和QM/ MM MD模拟方法的精确性和效率。
近年来基于分块的量子力学计算方法的发展为大的QM分区提供了可能177,178,并在QM/MM计
算中获得应用。利用大分子体系内的“化学局域性”,将较大的QM区域分块处理,在减少计算量的同时获取更为精确的结果,目前已经逐步应用于蛋白质-配体结合自由能和蛋白质NMR化学位移等问题的计算,在药物设计中具有重要应用价值178-181。粗粒化分子动力学能够在有限的计算能力下,对大尺度的分子体系进行描述,目前已经应用于生物膜及蛋白质生物合成等问题的计算中,在生物化学和分子生物学的理解中具有重要意义182,183,通过和QM/MM方法的结合,拓展多尺度模拟方法的应用。增强抽样方法的发展有助于改进QM/MM MD方法扫描蛋白质构象和预测其热力学性质的能力,使得理论与实验的直接可比性更强184-186。介观动力学的发展使宏观热力学与微观动力学有效结合,在药物运输等方面具有十分重要的应用价值187。总之,这些多尺度计算模拟方法的发展和结合,可以突破现有方法在体系大小、时间尺度、准确性等方面的局限,实现复杂生物体系和酶催化过程的多尺度全程模拟。
(1)Albery,W.J.;Knowles,J.R.Biochemistry 1976,15,5631. doi:10.1021/bi00670a032
(2) Chien,Y.H.;Lee,N.C.;Chiang,S.C.;Desnick,R.J.;Hwu, W.L.Mol.Med.2012,18,780.doi:10.2119/ molmed.2012.00002
(3) Fersht,A.Structure and Mechanism in Protein Science:a Guide to Enzyme Catalysis and Protein Folding;Freeman: New York,1999.
(4) Garcia-Viloca,M.;Gao,J.;Karplus,M.;Truhlar,D.G.Science 2004,303,186.doi:10.1126/science.1088172
(5) Macedo,M.F.;Quinta,R.;Pereira,C.S.;Miranda,M.C.S. Mol.Genet.Metab.2012,106,83.doi:10.1016/j. ymgme.2012.02.014
(6) Mulholland,A.J.Drug Discovery Today 2005,10,1393. doi:10.1016/S1359-6446(05)03611-1
(7) Schomburg,I.;Chang,A.;Placzek,S.;Söhngen,C.;Rother, M.;Lang,M.;Munaretto,C.;Ulas,S.;Stelzer,M.;Grote,A. Nucleic Acids Res.2012,gks1049.doi:10.1093/nar/gks1049
(8) Warshel,A.;Sharma,P.K.;Kato,M.;Yu,X.;Liu,H.;Olsson, M.H.M.Chem.Rev.2006,106,3210.doi:10.1002/ chin.200642255
(10) Huang,Y.S.;Zhang,G.Y.Biochemistry and Molecular Biology;Science Press:Beijing,2006.[黄诒森,张光毅.生物化学与分子生物学.北京:科学出版社,2006.]
(11)Wu,R.;Gong,W.;Liu,T.;Zhang,Y.;Cao,Z.J.Phys.Chem.B 2012,116,1984.doi:10.1021/jp211403j
(12)Wu,R.;Xie,H.;Cao,Z.;Mo,Y.J.Am.Chem.Soc.2008,130, 7022.doi:10.1021/ja710633c
(13)Xin,Z.;Yuan,Z.;Yan,H.;Cao,Z.;Mo,Y.J.Comput.Chem. 2016,37,1163.doi:10.1002/jcc.24306
(14)Wijeyewickrema,L.C.;Duncan,R.C.;Pike,R.N.Front. Immunol.2014,5,444.doi:10.3389/fimmu.2014.00444
(15)Chen,N.;Yuan,Z.;Lu,J.;Wu,R.;Cao,Z.J.Chem.Theory Comput.2015,11.doi:10.1021/acs.jctc.5b00045
(16)Zhao,Y.;Chen,N.;Mo,Y.;Cao,Z.Phys.Chem.Chem.Phys. 2014,16,26864.doi:10.1039/c4cp04032e
(17) Zhao,Y.;Chen,N.;Wang,C.;Cao,Z.ACS Catal.2016,6, 2145.doi:10.1021/acscatal.5b02855
(18)Zhao,Y.;Chen,N.;Wu,R.;Cao,Z.Phys.Chem.Chem.Phys. 2014,16,18406.doi:10.1039/c4cp01609b
(19)Warshel,A.;Sharma,P.K.;Kato,M.;Xiang,Y.;Liu,H.; Olsson,M.H.Chem.Rev.2006,106,3210.doi:10.1021/ cr0503106
(20) Carlsson,P.Biophys.J.2006,91,3151.doi:10.1529/ biophysj.106.082917
(21)And,T.W.;Duan,Y.J.Am.Chem.Soc.2007,129,6970. doi:10.1021/ja0691977
(22)Vashisth,H.;Abrams,C.F.Biophys.J.2008,95,4193. doi:10.1529/biophysj.108.139675
(23) Long,D.;Mu,Y.;Yang,D.Plos One 2009,4,e6081. doi:10.1371/journal.pone.0006081
(24) Peräkylä,M.Eur.Biophys.J.2009,38,185.doi:10.1007/ s00249-008-0369-x
(25) Klvana,M.;Pavlova,M.;Koudelakova,T.;Chaloupkova,R.; Dvorak,P.;Prokop,Z.;Stsiapanava,A.;Kuty,M.;Kuta-Smatanova,I.;Dohnalek,J.J.Mol.Biol.2009,392,1339. doi:10.1016/j.jmb.2009.06.076
(26) Pavlova,M.;Klvana,M.;Prokop,Z.;Chaloupkova,R.;Banas, P.;Otyepka,M.;Wade,R.C.;Tsuda,M.;Nagata,Y.; Damborsky,J.Nat.Chem.Biol.2009,5,727.doi:10.1038/ nchembio.205
(27)Wang,T.;Duan,Y.J.Mol.Biol.2009,392,1102.doi:10.1016/ j.jmb.2009.07.093
(28)Yao,L.;Li,Y.;Wu,Y.;Liu,A.;Yan,H.Biochemistry 2005,44, 5940.doi:10.1021/bi050095n
(29)Wagner,U.;Hasslacher,M.;Griengl,H.;Schwab,H.;Kratky,C. Structure 1996,4,811.doi:10.1016/S0969-2126(96)00088-3
(30) Gartler,G.;Kratky,C.;Gruber,K.J.Biotechnol.2007,129, 87.doi:10.1016/j.jbiotec.2006.12.009
(31) Darden,T.;Becker,O.;Mackerell,A.,Jr.;Roux,B.;Watanabe, M.;Becker,O.M.;MacKerell,A.D.;Jr.,Roux,B.;Watanabe, M.,Eds.2001,91.
(32) Frenkel,D.;Smit,B.Understanding Molecular Simulation: from Algorithms to Applications;Academic Press:New York 2001;Vol.1.
(33) Gruber,K.;Gartler,G.;Krammer,B.;Schwab,H.;Kratky,C. J.Biol.Chem.2004,279,20501.doi:10.1074/jbc.M401575200
(34) Case,D.;Darden,T.;Cheatham,T.E.,III.;Simmerling,C.; Wang,J.;Duke,R.;Luo,R.;Walker,R.;Zhang,W.;Merz,K. AMBER 12;University of California:San Francisco,2012.
(35) Phillips,J.C.;Braun,R.;Wang,W.;Gumbart,J.;Tajkhorshid, E.;Villa,E.;Chipot,C.;Skeel,R.D.;Kale,L.;Schulten,K.J. Comput.Chem.2005,26,1781.doi:10.1002/jcc.20289
(36) Berendsen,H.J.;van der Spoel,D.;van Drunen,R.Comput. Phys.Commun.1995,91,43.doi:10.1016/0010-4655(95) 00042-E
(37) Lindahl,E.;Hess,B.;Van Der Spoel,D.Mol.Model.Annu. 2001,7,306.doi:10.1007/s008940100045
(38) Hess,B.;Kutzner,C.;Van Der Spoel,D.;Lindahl,E.J.Chem. Theory Comput.2008,4,435.doi:10.1021/ct700301q
(39) Van Der Spoel,D.;Lindahl,E.;Hess,B.;Groenhof,G.;Mark, A.E.;Berendsen,H.J.J.Comput.Chem.2005,26,1701. doi:10.1002/jcc.20291
(40) Brooks,B.R.;Bruccoleri,R.E.;Olafson,B.D.;States,D.J.; Swaminathan,S.;Karplus,M.J.Comput.Chem.1983,4,187. doi:10.1002/jcc.540040211
(41) Ponder,J.W.TINKER,Software Tools for Molecular Design, Version 4.2;Washington University School of Medicine:Saint Louis,MO,2004.
(42)Plimpton,S.;Crozier,P.;Thompson,A.LAMMPS-Large-Scale Atomic/Molecular Massively Parallel Simulator;Sandia National Laboratories,2007,Vol.18.
(43) Warshel,A.;Levitt,M.J.Mol.Biol.1976,103,227. doi:10.1016/0022-2836(76)90311-9
(44) Cui,Q.;Karplus,M.J.Chem.Phys.2000,112,1133. doi:10.1063/1.480658
(45) Friesner,R.A.;Guallar,V.Annu.Rev.Phys.Chem.2005,56, 389.doi:10.1146/annurev.physchem.55.091602.094410
(46) Gao,J.;Amara,P.;Alhambra,C.;Field,M.J.J.Phys.Chem.A 1998,102,4714.doi:10.1021/jp9809890
(47)Hu,H.;Yang,W.Annu.Rev.Phys.Chem.2008,59,573. doi:10.1146/annurev.physchem.59.032607.093618
(48) Kerdcharoen,T.;Liedl,K.R.;Rode,B.M.Chem.Phys.1996, 211,313.doi:10.1016/0301-0104(96)00152-8
(49) Kouzarides,T.Cell 2007,128,693.doi:10.1016/j. cell.2007.02.005
(50) Lin,H.;Truhlar,D.G.Theor.Chem.Acc.2007,117,185. doi:10.1007/s00214-006-0143-z
(51)Lyne,P.D.;Hodoscek,M.;Karplus,M.J.Phys.Chem.A 1999,103,3462.doi:10.1021/jp982115j
(52) Mo,Y.;Gao,J.J.Comput.Chem.2000,21,1458.doi:10.1002/ 1096-987X(200012)21:16〈1458::AID-JCC4>3.0.CO;2-2
(53) Murphy,R.;Philipp,D.;Friesner,R.Chem.Phys.Lett.2000, 321,113.doi:10.1016/S0009-2614(00)00289-X
(54) Murphy,R.B.;Philipp,D.M.;Friesner,R.A.J.Comput. Chem.2000,21,1442.doi:10.1002/1096-987X(200012)21: 16〈1442::AID-JCC3>3.0.CO;2-O
(55) Senn,H.M.;Thiel,W.Top.Curr.Chem.2007,268,173. doi:10.1007/128_2006_084
(56) Senn,H.M.;Thiel,W.Curr.Opin.Chem.Biol.2007,11,182. doi:10.1016/j.cbpa.2007.01.684
(57) Senn,H.M.;Thiel,W.Angew.Chem.Int.Ed.2009,48,1198. doi:10.1002/anie.200802019
(58)Vreven,T.;Morokuma,K.;Farkas,Ö.;Schlegel,H.B.;Frisch, M.J.J.Comput.Chem.2003,24,760.doi:10.1002/jcc.10156
(59)Woodcock,H.L.;Hodošček,M.;Gilbert,A.T.;Gill,P.M.; Schaefer,H.F.;Brooks,B.R.J.Comput.Chem.2007,28, 1485.doi:10.1002/jcc.20587
(60) Zhang,Y.Theor.Chem.Acc.2006,116,43.doi:10.1007/ s00214-005-0008-x
(61)Maseras,F.;Morokuma,K.J.Comput.Chem.1995,16,1170. doi:10.1002/jcc.540160911
(62)Vreven,T.;Byun,K.S.;Komáromi,I.;Dapprich,S.; Montgomery,J.A.;Morokuma,K.;Frisch,M.J.J.Chem. Theory Comput.2006,2,815.doi:10.1021/ct050289g
(63) Geerke,D.P.;Thiel,S.;Thiel,W.;van Gunsteren,W.F.J. Chem.Theory Comput.2007,3,1499.doi:10.1021/ct7000123
(64) Elking,D.;Darden,T.;Woods,R.J.J.Comput.Chem.2007, 28,1261.doi:10.1002/jcc.20574
(65) Chen,J.;Martínez,T.J.Chem.Phys.Lett.2007,438,315. doi:10.1016/j.cplett.2007.02.065
(66) Kuo,I.F.W.;Mundy,C.J.;McGrath,M.J.;Siepmann,J.I.; VandeVondele,J.;Sprik,M.;Hutter,J.;Chen,B.;Klein,M.L.; Mohamed,F.J.Phys.Chem.B 2004,108,12990.doi:10.1021/ jp047788i
(67)Amara,P.;Field,M.J.Theor.Chem.Acc.2003,109,43. doi:10.1007/s00214-002-0413-3
(68) Das,D.;Eurenius,K.P.;Billings,E.M.;Sherwood,P.; Chatfield,D.C.;Hodošček,M.;Brooks,B.R.J.Chem.Phys. 2002,117,10534.doi:10.1063/1.1520134
(69) Eurenius,K.P.;Chatfield,D.C.;Brooks,B.R.;Hodoscek,M. Int.J.Quantum Chem.1996,60,1189.doi:10.1002/(SICI) 1097-461X(1996)60:6〈1189::AID-QUA7>3.0.CO;2-W
(70) Ferré,N.;Olivucci,M.J.Mol.Struct.:Theochem 2003,632, 71.doi:10.1016/S0166-1280(03)00289-6
(71)Singh,U.C.;Kollman,P.A.J.Comput.Chem.1986,7,718. doi:10.1002/jcc.540070604
(72) Field,M.J.;Bash,P.A.;Karplus,M.J.Comput.Chem.2004, 11,700.doi:10.1002/jcc.540110605
(73) Zhang,Y.;Liu,H.;Yang,W.J.Chem.Phys.2000,112,3483. doi:10.1063/1.480503
(74) Zhang,Y.J.Chem.Phys.2005,122,024114.doi:10.1063/ 1.1834899
(75)Antes,I.;Thiel,W.J.Phys.Chem.A 1999,103,9290. doi:10.1021/jp991771w
(76) Poteau,R.;Ortega,I.;Alary,F.;Solis,A.R.;Barthelat,J.C.; Daudey,J.P.J.Phys.Chem.A 2001,105,198.doi:10.1021/jp002500k
(77)Yasuda,K.;Yamaki,D.J.Chem.Phys.2004,121,3964. doi:10.1063/1.1772354
(78) DiLabio,G.A.;Hurley,M.M.;Christiansen,P.A.J.Chem. Phys.2002,116,9578.doi:10.1063/1.1477182
(79) Kairys,V.;Jensen,J.H.J.Phys.Chem.A 2000,104,6656. doi:10.1021/jp000887l
(80)Assfeld,X.;Rivail,J.L.Chem.Phys.Lett.1996,263,100.doi: 10.1016/S0009-2614(96)01165-7
(81) Ferré,N.;Assfeld,X.;Rivail,J.L.J.Comput.Chem.2002,23, 610.doi:10.1002/jcc.10058
(82) Monard,G.;Loos,M.;Théry,V.;Baka,K.;Rivail,J.L.Int.J. Quantum Chem.1996,58,153.doi:10.1002/(SICI)1097-461X (1996)58:2〈153::AID-QUA4>3.0.CO;2-X
(83) Pu,J.;Gao,J.;Truhlar,D.G.J.Phys.Chem.A 2004,108,632. doi:10.1021/jp036755k
(84) Théry,V.;Rinaldi,D.;Rivail,J.L.;Maigret,B.;Ferenczy,G. G.J.Comput.Chem.1994,15,269.doi:10.1002/ jcc.540150303
(85) Zhang,Y.;Lee,T.S.;Yang,W.J.Chem.Phys.1999,110,46. doi:10.1063/1.478083
(86)Alary,F.;Poteau,R.;Heully,J.L.;Barthelat,J.C.;Daudey,J. P.Theor.Chem.Acc.2000,104,174.doi:10.1007/ s002140000138
(87) Bessac,F.;Alary,F.;Carissan,Y.;Heully,J.L.;Daudey,J.P.; Poteau,R.J.Mol.Struct.:Theochem 2003,632,43. doi:10.1016/S0166-1280(03)00287-2
(88) Carissan,Y.;Bessac,F.;Alary,F.;Heully,J.L.;Poteau,R.Int. J.Quantum Chem.2006,106,727.doi:10.1002/qua.20837
(89)DiLabio,G.A.;Wolkow,R.A.;Johnson,E.R.J.Chem.Phys. 2005,122,044708.doi:10.1063/1.1839857
(90) Slavíček,P.;Martínez,T.J.J.Chem.Phys.2006,124,084107. doi:10.1063/1.2173992
(91)Gao,J.;Thompson,M.A.Combined Quantum Mechanical and Molecular Mechanical Methods;American Chemical Society:Washington,DC,1998;Vol.712.
(92) Philipp,D.M.;Friesner,R.A.J.Comput.Chem.1999,20, 1468.doi:10.1002/(SICI)1096-987X(19991115)20:143.0.CO; 2-0
(93)Amara,P.;Field,M.J.;Alhambra,C.;Gao,J.Theor.Chem. Acc.2000,104,336.doi:10.1007/s002140000153
(94) Garcia-Viloca,M.;Gao,J.Theor.Chem.Acc.2004,111,280. doi:10.1007/s00214-003-0512-9
(95) Pu,J.;Gao,J.;Truhlar,D.G.ChemPhysChem 2005,6,1853. doi:10.1002/cphc.200400602
(96) Jensen,J.H.;Day,P.N.;Gordon,M.S.;Basch,H.;Cohen,D.; Garmer,D.R.;Kraus,M.;Stevens,W.J.ACS Symp.Ser.1994, 569,139.doi:10.1021/bk-1994-0569.ch009
(97) Day,P.N.;Jensen,J.H.;Gordon,M.S.;Webb,S.P.;Stevens, W.J.;Krauss,M.;Garmer,D.;Basch,H.;Cohen,D.J.Chem. Phys.1996,105,1968.doi:10.1063/1.472045
(98) Gordon,M.S.;Freitag,M.A.;Bandyopadhyay,P.;Jensen,J. H.;Kairys,V.;Stevens,W.J.J.Phys.Chem.A 2001,105,293. doi:10.1021/jp002747h
(99) Kumar,S.;Rosenberg,J.M.;Bouzida,D.;Swendsen,R.H.; Kollman,P.A.J.Comput.Chem.1992,13,1011.doi:10.1002/ jcc.540130812
(100)Souaille,M.;Roux,B.T.Comput.Phys.Commun.2001,135, 40.doi:10.1016/S0010-4655(00)00215-0
(101) Ferrenberg,A.M.;Swendsen,R.H.Phys.Rev.Lett.1988,61, 2635.doi:10.1103/PhysRevLett.61.2635
(102) Grossfield,A.WHAM:the Weighted Histogram Analysis Method,version 2.0.6;Grossfield Lab:Rochester,NY,2012. http://membrane.urmc.rochester.edu/content/wham
(103) Zwanzig,R.W.J.Chem.Phys.1954,22,1420.doi:10.1063/ 1.1740409
(104) Kollman,P.Chem.Rev.1993,93.doi:10.1021/cr00023a004
(105) Jorgensen,W.L.Acc.Chem.Res.1989,22,184.doi:10.1021/ ar00161a004
(106) Kirkwood,J.G.J.Chem.Phys.1935,3,300.doi:10.1063/ 1.1749657
(107) Laio,A.;Parrinello,M.Proc.Natl.Acad.Sci.2002,99,12562. doi:10.1073/pnas.202427399
(108) Laio,A.;Rodriguezfortea,A.;Gervasio,F.L.;Ceccarelli,M.; Parrinello,M.J.Phys.Chem.B 2005,109,6714.doi:10.1021/ jp045424k
(109) Iannuzzi,M.;Laio,A.;Parrinello,M.Phys.Rev.Lett.2003,90, 238302.doi:10.1103/PhysRevLett.90.238302
(110) Jarzynski,C.Phys.Rev.Lett.1996,78,2690.doi:10.1103/ PhysRevLett.78.2690
(111) Park,S.;Khaliliaraghi,F.;Tajkhorshid,E.;Schulten,K.J. Chem.Phys.2003,119,3559.doi:10.1063/1.1590311
(112) Park,S.;Schulten,K.J.Chem.Phys.2004,120,5946. doi:10.1063/1.1651473
(113) Crooks,G.E.Phys.Rev.E:Stat.Phys.Plasmas Fluids 2000, 61,2361.doi:10.1103/PhysRevE.61.2361
(114) Bonomi,M.;Branduardi,D.;Bussi,G.;Camilloni,C.;Provasi, D.;Raiteri,P.;Donadio,D.;Marinelli,F.;Pietrucci,F.; Broglia,R.A.Comput.Phys.Commun.2009,180,1961. doi:10.1016/j.cpc.2009.05.011
(115) Piccinini,E.;Ceccarelli,M.;Affinito,F.;Brunetti,R.; Jacoboni,C.J.Chem.Theory Comput.2008,4,173. doi:10.1021/ct7001896
(116) Kästner,J.WIREs Comput.Mol.Sci.2011,1,932.doi: 10.1002/wcms.66
(117) Radmer,R.J.;Kollman,P.A.J.Comput.Chem.1997,18,902. doi:10.1002/(SICI)1096-987X(199705)18:7〈902::AID-JCC4>3.0.CO;2-V
(118) Jorgensen,W.L.;Thomas,L.L.J.Chem.Theory Comput. 2008,4,869.doi:10.1021/ct800011m
(119)Aaqvist,J.J.Phys.Chem.1990,94,8021.doi:10.1021/ j100384a009
(120) Darve,E.;Pohorille,A.J.Chem.Phys.2001,115,9169. doi:10.1063/1.1410978
(121) Hénin,J.;Chipot,C.J.Chem.Phys.2004,121,2904. doi:10.1063/1.1773132
(122) Comer,J.;Gumbart,J.C.;Hénin,J.;Lelièvre,T.;Pohorille,A.; Chipot,C.J.Phys.Chem.B 2014,119,1129.doi:10.1021/ jp506633n
(123) Provasi,D.;Bortolato,A.;Filizola,M.Biochemistry 2009,48, 10020.doi:10.1021/bi901494n
(124) Zhang,Z.S.;Wang,Q.;Chen,E.Y.Sci.China Chem.2014, 44,854.[张志森,王 琦,陈尔余.中国科学:化学,2014,44, 854.]doi:10.1360/N032014-00005
(125) Hou,T.;Guo,S.;Xu,X.J.Phys.Chem.B 2002,106,5527. doi:10.1021/jp015516z
(126) Lepšík,M.;Kříž,Z.;Havlas,Z.Proteins:Struct.,Funct., Bioinf.2004,57,279.doi:10.1002/prot.20192
(127)Brown,S.P.;Muchmore,S.W.J.Chem.Inf.Comput.Sci. 2006,46,999.doi:10.1002/chin.200630220
(128)Alcaro,S.;Artese,A.;Ceccherini-Silberstein,F.;Ortuso,F.; Perno,C.F.;Sing,T.;Svicher,V.J.Chem.Inf.Comput.Sci. 2009,49,1751.doi:10.1021/ci900012k
(129) Hou,T.;Yu,R.J.Med.Chem.2007,50,1177.doi:10.1021/ jm0609162
(130) Stoica,I.;Sadiq,S.K.;Coveney,P.V.J.Am.Chem.Soc.2008, 130,2639.doi:10.1021/ja0779250
(131) Wang,W.;Kollman,P.A.J.Mol.Biol.2000,303,567. doi:10.1006/jmbi.2000.4057
(132)Gohlke,H.;Case,D.A.J.Comput.Chem.2004,25,238. doi:10.1002/jcc.10379
(133)Hou,T.;Chen,K.;McLaughlin,W.A.;Lu,B.;Wang,W.PLoS Comput.Biol.2006,2,e1.doi:10.1371/journal.pcbi.0020001
(134) Pearlman,D.A.J.Med.Chem.2005,48,7796.doi:10.1021/ jm050306m
(135) Kuhn,B.;Gerber,P.;Schulz-Gasch,T.;Stahl,M.J.Med. Chem.2005,48,4040.doi:10.1021/jm049081q
(136) Hou,T.;Li,N.;Li,Y.;Wang,W.J.Proteome Res.2012,11, 2982.doi:10.1021/pr3000688
(137) Hickel,A.;Hasslacher,M.;Griengl,H.Physiol.Plant.1996, 98,891.doi:10.1111/j.1399-3054.1996.tb06700.x
(138) Lieberei,R.;Selmar,D.;Biehl,B.Plant Syst.Evol.1985,150, 49.doi:10.1007/BF00985567
(139) Gruber-Khadjawi,M.;Purkarthofer,T.;Skranc,W.;Griengl, H.Adv.Synth.Catal.2007,349,1445.doi:10.1002/ chin.200740040
(140) Oroz-Guinea,I.;García-Junceda,E.Curr.Opin.Chem.Biol. 2013,17,236.doi:10.1016/j.cbpa.2013.02.015
(141) Müller,M.ChemBioEng Rev.2014,1,14.doi:10.1002/ cben.201300005
(142) Diebler,J.;von Langermann,J.;Mell,A.;Hein,M.;Langer,P.; Kragl,U.ChemCatChem 2014,6,987.doi:10.1002/ cctc.201300965
(143) Purkarthofer,T.;Skranc,W.;Schuster,C.;Griengl,H.Appl. Microbiol.Biotechnol.2007,76,309.doi:10.1007/s00253-007-1025-6
(144) Holt,J.;Hanefeld,U.ChemInform 2009,40,15.doi:10.1002/ chin.200940251
(145) Turner,N.J.;O'Reilly,E.Nat.Chem.Biol.2013,9,285. doi:10.1038/nchembio.1235
(146) Hanefeld,U.Chem.Soc.Rev.2013,42,6308.doi:10.1039/ C3CS35491A
(147)Sharma,M.;Sharma,N.N.;Bhalla,T.C.Enzyme Microb. Technol.2005,37,279.doi:10.1016/j.enzmictec.2005.04.013
(148) Dadashipour,M.;Asano,Y.ACS Catal.2011,1,1121. doi:10.1021/cs200325q
(149) Dreveny,I.;Andryushkova,A.S.;Glieder,A.;Gruber,K.; Kratky,C.Biochemistry 2009,48,3370.doi:10.1021/ bi802162s
(150) Versées,W.;Steyaert,J.Curr.Opin.Struct.Biol.2003,13, 731.doi:10.1016/j.sbi.2003.10.002
(151)Downie,M.J.;Kirk,K.;Mamoun,C.B.Eukaryotic Cell 2008, 7,1231.doi:10.1128/EC.00159-08
(152) Miles,R.W.;Tyler,P.C.;Evans,G.B.;Furneaux,R.H.; Parkin,D.W.;Schramm,V.L.Biochemistry 1999,38,13147. doi:10.1021/bi990829u
(153) Ranquin,A.;Versées,W.;Meier,W.;Steyaert,J.;Van,G.P. Nano Lett.2005,5,2220.doi:10.1021/nl051523d
(154) Parkin,D.W.J.Biol.Chem.1996,271,21713.doi:10.1074/ jbc.271.36.21713
(155)Vandemeulebroucke,A.;Versées,W.;De Vos,S.;Van Holsbeke,E.;Steyaert,J.Biochemistry 2003,42,12902. doi:10.1021/bi0347914
(156) Vandemeulebroucke,A.;Versées,W.;Jan Steyaert,A.;Barlow, J.N.Biochemistry 2006,45,9307.doi:10.1021/bi060666r
(157) Barlow,J.N.;Steyaert,J.Biochim.Biophys.Acta 2007,1774, 1451.doi:10.1016/j.bbapap.2007.08.027
(158) Versées,W.;Loverix,S.;An,V.;Geerlings,P.;Steyaert,J.J. Mol.Biol.2004,338,1.doi:10.1016/j.jmb.2004.02.049
(159) Chen,N.;Hu,G.;Xu,J.;Cao,Z.;Wu,R.Biochim.Biophys. Acta 2013,1834,1117.doi:10.1016/j.bbapap.2013.02.005
(160) Warren,L.;Gottschalk,A.Glycoproteins,Their Composition, Structure and Function;Elsevier:Amsterdam,197;p 1097.
(161) Vincent,F.;Davies,G.J.;Brannigan,J.A.J.Biol.Chem. 2005,280,19649.doi:10.1074/jbc.M502131200
(162) Liu,C.;Li,D.;Liang,Y.H.;Li,L.F.;Su,X.D.J.Mol.Biol. 2008,379,73.doi:10.1016/j.jmb.2008.03.031
(163) Álvarez-Añorve,L.I.;Bustos-Jaimes,I.;Calcagno,M.L.; Plumbridge,J.J.Bacteriol.2009,191,6401.doi:10.1128/ JB.00633-09
(164) Bustos-Jaimes,I.;Sosa-Peinado,A.;Rudiño-Piñera,E.; Horjales,E.;Calcagno,M.L.J.Mol.Biol.2002,319,183. doi:10.1016/S0022-2836(02)00096-7
(165)Uhde,A.;Youn,J.W.;Maeda,T.;Clermont,L.;Matano,C.;Krämer,R.;Wendisch,V.F.;Seibold,G.M.;Marin,K.Appl. Microbiol.Biotechnol.2013,97,1679.doi:10.1007/s00253-012-4313-8
(166)Kawada-Matsuo,M.;Mazda,Y.;Oogai,Y.;Kajiya,M.;Kawai, T.;Yamada,S.;Miyawaki,S.;Oho,T.;Komatsuzawa,H.Plos One 2012,7,e33382.doi:10.1371/journal.pone.0033382
(167) Liu,L.;Liu,Y.;Shin,H.D.;Chen,R.;Li,J.;Du,G.;Chen,J. Appl.Microbiol.Biotechnol.2013,97,6149.doi:10.1007/ s00253-013-4995-6
(168)Dong,L.;Su-Ying,M.A.;Yang,L.M.;Sun,X.D.Lishizhen Medicine and Materia Medica Research 2008,19,836.[董丽,马素英,杨利敏,孙祥德.时珍国医国药,2008,19,836.] doi:1008-0805(2008)04-0836-02
(169) Rudino-Pinera,E.;Morales-Arrieta,S.;Rojas-Trejo,S.; Horjales,E.Acta Crystallogr.Sect.D.Biol.Crystallogr.2001, 58,10.doi:10.1107/S0907444901016699
(170) Bühler,H.;Effenberger,F.;Förster,S.;Roos,J.;Priv.-Doz.,H. W.ChemBioChem 2003,4,211.doi:10.1002/cbic.200390033
(171) Bühler,H.;Miehlich,B.;Effenberger,F.ChemBioChem 2005, 6,711.doi:10.1002/cbic.200400302
(172) Zhang,R.;Monsma,F.Expert Opin.Drug Discovery 2010,5, 1023.doi:10.1517/17460441.2010.520700
(173) Xu,W.F.Drug Design;People′s Medical Publishing House: Beijing,2007.[徐文方.药物设计学.北京:人民卫生出版社, 2007.]
(174) Chen,K.X.;Jiang,H.L.;Ji,R.Y.Computer Aided Drug Design,Principle,Method,and Application;Shanghai Scientific&Technical Publishers:Shanghai,2000.[陈凯先,蒋华良,嵇汝运.计算机辅助药物设计:原理、方法及应用.上海:上海科学技术出版社,2000.]
(175) Ma,J.A.;Huang,R.Q.Chin.J.Synth.Chem.2000,8,479.[马军安,黄润秋.合成化学,2000,8,479.]doi:10.3969/j. issn.1005-1511.2000.06.004
(176) Zhou,J.;Li,M.;Chen,N.;Wang,S.;Luo,H.B.;Zhang,Y.; Wu,R.ACS Chem.Biol.2015,10,687.doi:10.1021/ cb500767c
(177) Li,S.;Li,W.;Ma,J.Acc.Chem.Res.2014,47,2712. doi:10.1021/ar500038z
(178) He,X.;Zhu,T.;Wang,X.;Liu,J.;Zhang,J.Z.Acc.Chem. Res.2014,47,2748.doi:10.1021/ar500077t
(179)Wang,B.;Yang,K.R.;Xu,X.;Isegawa,M.;Leverentz,H.R.; Truhlar,D.G.Acc.Chem.Res.2014,47,2731.doi:10.1021/ ar500068a
(180) Pruitt,S.R.;Bertoni,C.;Brorsen,K.R.;Gordon,M.S.Acc. Chem.Res.2014,47,2786.doi:10.1021/ar500097m
(181) Rao,L.;Chi,B.;Ren,Y.;Li,Y.;Xu,X.;Wan,J.J.Comput. Chem.2016,37,336 doi:10.1002/jcc.24217
(182) Zhang,L.;Pardo-Avila,F.T.;Unarta,I.C.;Cheung,P.P.H.; Wang,G.;Wang,D.;Huang,X.Acc.Chem.Res.2016,49, 687.doi:10.1021/acs.accounts.5b00536
(183) Bond,P.J.;Sansom,M.S.J.Am.Chem.Soc.2006,128,2697. doi:10.1021/ja0569104
(184)Gao,Y.Q.;Yang,L.J.Chem.Phys.2006,125,114103. doi:10.1063/1.2244548
(185) Yang,L.;Shao,Q.;Gao,Y.Q.J.Chem.Phys.2009,130, 124111.doi:10.1063/1.3097129
(186) Zhang,J.;Yang,Y.I.;Yang,L.;Gao,Y.Q.J.Phys.Chem.B 2015,119,5518.doi:10.1021/jp511057f
(187)Dai,X.;Ding,H.;Yin,Q.;Wan,G.;Shi,X.;Qiao,Y.J.Mol. Graphics Modell.2015,57,20.doi:10.1016/j. jmgm.2015.01.002
Global Simulations of Enzymatic Catalysis
ZHAO Yuan1CAO Ze-Xing2,*
(1KeyLaboratoryofNaturalMedicineandImmuno-Engineering,HenanUniversity,Kaifeng475004,HenanProvince,P.R.China;2Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 360015,Fujian Province,P.R.China)
Enzymatic catalytic processes generally involve substrate delivery,selective catalytic reaction,and product release.Owing to the complex protein environment effect,any nonchemical or chemical step may determine the enzyme activity.Herein,to comprehensively understand enzymatic activity,extensive combined quantum mechanics/molecular mechanics(QM/MM)and molecular mechanics(MM)molecular dynamics(MD) simulations were carried out on several kinds of enzymes.Possible reaction mechanisms,roles of the conserved residues,and effects of the protein environment on the whole enzymatic process are discussed in detail,which will enrich the knowledge of reactivity in proteins.With the improvement and development of multiscale models and computational methods,it is expected that global simulations of extremely large and complicated enzymes will enable and lend support to enzyme engineering.
Enzymatic catalysis;Substrate delivery;Free energy calculations;QM/MM MD simulation; Random acceleration molecular dynamics(DAMD)simulation
O641
Zhang,X.;Houk,K.Acc.Chem.Res.2005,38,379.
10.1021/ar040257s
doi:10.3866/PKU.WHXB201612191
Received:October 28,2016;Revised:December 19,2016;Published online:December 19,2016.
*Corresponding author.Email:zxcao@xmu.edu.cn;Tel:+86-592-2186081.
The project was supported by the National Natural Science Foundation of China(21133007,21373164,21172053).国家自然科学基金(21133007,21373164,21172053)资助项目
