一种新型的蛋白质复合物挖掘算法
2017-04-11许睿金松林王廷雨
许睿,金松林,王廷雨
(河南科技学院信息工程学院,河南新乡453003)
一种新型的蛋白质复合物挖掘算法
许睿,金松林,王廷雨
(河南科技学院信息工程学院,河南新乡453003)
随着人类基因组计划的完成,如何从蛋白质网络的结构特性出发,有效地识别蛋白质复合物和功能模块正成为蛋白质组学研究的重点.提出一种新型的蛋白质复合物挖掘算法,首先分析蛋白质网络的结构特征,依据每个蛋白质的GO术语相似性对蛋白质网络进行加权,再通过不断地迭代传播,将各节点划分到稳定的集合中,从而识别出蛋白质复合物.该算法与CPM算法(k=3)及Core-Attachment算法进行对照实验,结果表明该算法在预测得到的蛋白质复合物的匹配程度和复合物的功能富集性等方面更有优势.
蛋白质网络;GO术语;蛋白质相互作用;蛋白质复合物
随着人类基因组序列测序的完成,现代生物学研究已经进入了后基因时代,课题研究的重点也将转移到基因的功能表达问题,大多数基因最终是通过对应的蛋白质进行表达的[1].在蛋白质组学的大量研究中,发现一个复杂的生物活动不可能只是单独的生物分子来操纵的,往往是由多个生物分子相互配合所表达出来的,如何透过生命现象,观察生命的本质以及发现基因的全部功能是一个重要的挑战[2].
研究表明蛋白质网络中各节点的度服从幂率分布[3],大部分节点的度较低而少数节点的度却很高,而且大部分节点只与少数节点有相互作用,少数节点却与多数节点有相互作用.具体的蛋白质网络在实现某种功能的过程中,节点间的交互是有条不紊地进行的.研究表明蛋白质网络中存在着簇结构,该簇结构是由具有相似功能的蛋白质节点组成,这些节点间的交互程度明显高于非簇结构的节点间的交互程度,蛋白质网络中大部分生命活动都是由多个簇之间的相互作用来完成[4].由于生物系统的功能复杂,通过挖掘蛋白质网络中的簇结构,有助于了解蛋白质网络的结构和功能特性.
1 相关定义说明
1.1基因本体(Gene Ontology)
基因本体是“基因本体联盟”所建立的数据库.基因本体使各种数据库中基因产物功能描述相一致,统一了各种数据库中关于基因的定义,使研究者能够进行统一的归纳、处理、共享基因以及基因产物的数据.基因本体包括分子功能(molecular function)、生物学过程(biological process)、细胞组件(cellular component).分子功能描述在个体分子生物学上的活性,如催化活性或结合活性.生物学过程是由分子功能有序地组成并具有多个步骤的一个过程.细胞组件指细胞的每个部分和细胞外环境,揭示了基因产物是在什么地方起作用.
GO术语的结构如图1所示,是一个有向无环图.节点表示术语,边表示两术语间的关系,其中“I”表示继承关系,“P”表示从属关系,“R”表示前者调节控制后者.如果一个节点术语A被另一个节点术语B所包含或者继承,则称节点A为节点B的父节点,节点B为节点A的子节点.在图1中,从上而下,GO术语语义逐渐具体,子节点拥有其祖先节点的注释信息,因此下层节点所拥有的语义信息量比父节点要更大.

图1GO术语结构Fig.1 Structure diagramofGOterms
1.2 P-value值
在蛋白质网络中,处在同一蛋白质功能模块内部的蛋白质往往具有相似的结构和功能特性,它们之间相互作用,共同实现某种生物功能.通过计算具有共同功能的蛋白质在预测蛋白质复合物中出现的概率的P-value值,来判断蛋白质功能模块的主要功能或者预测新功能.P-value值的定义如式(1)所示
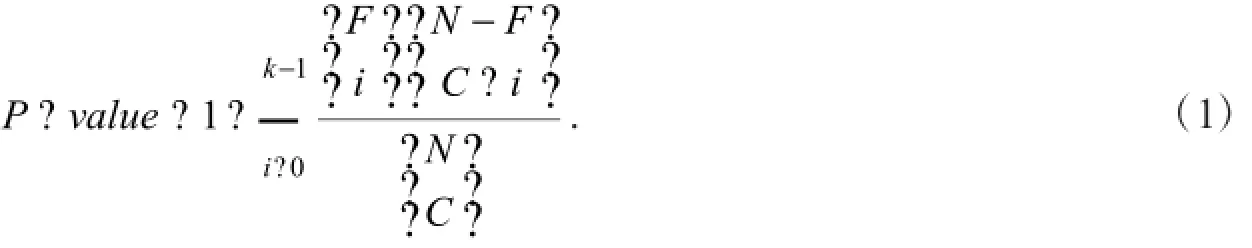
式(1)中,N表示蛋白质网络的节点数,C表示蛋白质复合物中的蛋白质数,k表示蛋白复合物中具有某个功能的蛋白质数,F表示蛋白质网络的全部拥有该功能的蛋白质数.一般认为在蛋白质网络中, P-value值越低,蛋白质复合物随机出现这种功能的可能性就越低,因此这样预测蛋白质复合物具有更高的统计意义.通常将预测蛋白质复合物对应P-value值最小的功能作为其主要功能或者注释功能.
2 衡量蛋白质相似性及算法描述
2.1 GO术语的相似性
蛋白质复合物的GO语义相似性是指所有蛋白质间的平均关联程度,可以通过所有蛋白质复合物的加权平均计算实现.通常,蛋白质复合物具有较高的GO语义相似性,表明复合物内的蛋白质表达相似功能的概率越大.在计算两个GO术语间的相似度的时候,如果两个GO术语共享的信息越多,则两者就越相似.在本文中,采用Lin[5]提出的计算方法,两个GO术语的相似性由它们所共有的最近祖先的信息量和每个GO术语包含的信息量共同决定.式(2)如下所示

式(2)中,C1和C2分别表示两个GO术语,p(c)表示该术语C在数据集中出现的概率,CT(C1,C2)表示C1、C2共同祖先的集合.
2.2 蛋白质相互作用
通过研究发现,在蛋白质网络中,每一个蛋白质都有多条GO注释信息,本文计算两个蛋白质之间相似性的最大值来衡量这两者的相互作用.因此两个蛋白质之间的蛋白质相互作用E(u,v)可以用蛋白质u、蛋白质v的GO语义相似来衡量,如式(3)所示

式(3)中,Fu表示蛋白质u的GO注释集,Fv表示蛋白质v的GO注释集.文本用蛋白质间相似性来衡量两个蛋白质之间的相互作用,将蛋白质网络从无权图转化为有权图,在转化的过程中,用GO术语作为参考,突出了蛋白质之间的生物学特性.
2.3 算法描述
在蛋白质网络中,用两蛋白质间的GO术语的相似性强弱量化这两者间的相互作用,进而把无权的蛋白质网络转化为有权网.将初始时刻每个蛋白质节点当作单独的蛋白质模块,由于每个模块只含有一个节点,则模块的唯一信号就由该节点来表示.本算法设定模块间的信号只允许在直接连接的相邻节点间进行传播,各节点依据相互作用关系将自己的信号传到相邻节点集合中,并且该节点也能收到相邻节点集反馈给自己的信号,而且每个蛋白质节点在传播过程中都会将信号强度最大的信号类型来替换自己原先的类型.按照这一设定,经过一系列迭代过程,蛋白质网络中会形成一些节点集合,通过分析,发现每个节点集合中的节点具有相同的信号类型,而且他们在结构上是紧密连接的.随着迭代过程的不断进行,节点集合会将具有结构相似以及信号类型一致的单独节点加入其中.当整个蛋白质网络中的各节点集合的信号类型趋于稳定的时候,算法终止.最后,分析各蛋白质节点的信号类型,具有相同的信号类型的节点预测会具有相似的功能特性,划为同一个蛋白质复合物中.
算法的具体描述如下:

为每个节点分配唯一的信号类型
计算该节点的相邻节点的信号量
用最强相邻节点的信号类型替换自己原有的信号类型
until蛋白质网络中的信号类型稳定
f
or所有信号类型
输出具有该信号类型的节点
end for
3 实验与分析
通过分析所有生物物种的蛋白质相互作用的数据,发现酵母蛋白质网络的数据较为完备,所以本文选择酵母蛋白质网络作为本实验的测试对象.实验选取Krogan数据库[6],先对数据集进行预处理,过滤掉自环边和多边作用等,最终得到的测试网络包括了3 672个节点和14 317条边.为了验证本算法的有效性,将实验结果与另两个基于稠密子图的蛋白质复合物挖掘算法(CPM[7]、Core-Attachment[8])的实验结果做对照分析.为了有效地评价预测得到的蛋白质复合物,从已发布的小规模实验数据[9]中人工提取了408个蛋白质复合物并生成了详细目录.形成该目录的标准是复合物的规模大于3,选取了236个标准复合物,其平均规模为6.6.

表1 参数P在不同值下的聚类结果Tab.1 Clusteringresult in different value ofparameter P
在表1中,当衰减因子P值不断增大时,本算法识别的蛋白质复合物的个数随之增加,尤其在P值从0到0.2这一阶段,识别得到的复合物数量增长最为迅速.同时蛋白质复合物的平均规模在不断减小,复合物中所包含的节点数量也在不断减小.在P值为0时,在蛋白质网络中形成节点规模很大的蛋白质复合物.研究表明,当匹配阈值设定为0.2时,就可以将此时识别的蛋白质复合物标记为已知蛋白质复合物.但是为了找到最适合酵母蛋白质网络的参数P值,将参数P设置从0到1,步长为0.1,进行算法有效性分析.统计本算法在不同参数P的条件下,识别出的蛋白质复合物在已知复合物中所占的比例,如图2所示.
在图2中,通过分析,发现本算法在参数P为0.4时,预测准确率最高,也就是预测得到的蛋白质复合物标识正确的比例最高.因此,本实验将参数P设定为0.4.Core-Attachment算法、CPM算法(k=3)和本算法分别预测得到的蛋白质复合物被标识的比例见图3.
从图3中可以看出,在不同匹配阈值下,本算法预测得到的复合物被标识的比例都要略高于CPM算法(k=3)得到的结果,而且明显高于Core-Attachment算法得到的结果.一般认为当匹配阈值大于0.2时,可以将此时识别的蛋白质复合物标记为已知蛋白质复合物.本算法预测到的蛋白质复合物中有约40%的匹配阈值大于0.2,而CPM算法(k=3)和Core-Attachment算法预测得到的蛋白质复合物匹配阈值大于0.2复合物的比例分别为38%和18%,本算法比其他两种算法分别提高了2个和22个百分点.
对本算法、CPM算法(k=3)和Core-Attachment算法预测得到的蛋白质复合物分别进行功能富集性分析.比较这3种不同算法得到的蛋白质复合物的P-value分布,将P-value按(-∞,E-10)、[E-10,E-5)、[E-5,0.01)、[0.01,+∞)的从小到大分为4个区间,分别统计各区间上蛋白质复合物的数量与比例,各算法的P-value分布如表2所示.
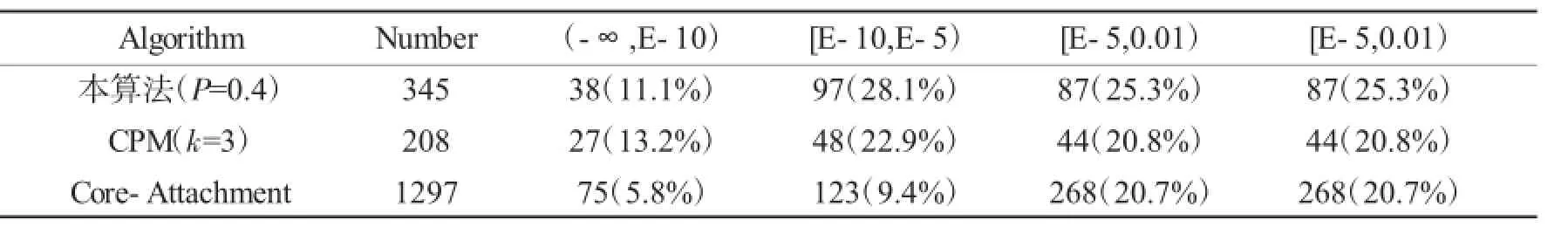
表2 不同算法识别的蛋白质复合物的功能富集性分析Tab.2 Functional enrichment ofprotein complexes predicted bydifferent algorithms
研究表明,当P-value<0.01时,所对应的蛋白质复合物功能表现更加明显,具有生物学意义.从表2中可以看出,本算法预测得到的蛋白质复合物中有约64.5%的复合物具有相应的生物学意义,高于CPM算法(56.9%)和Core-Attachment算法(35.7%).本算法在P-value值小于E-10时所占蛋白质复合物比例达到11.1%,比Core-Attachment算法对应的比值提高近一倍,比CPM算法略低.但是在P-value值在[E-10,E-5)和[E-5,0.01)时,本算法得到的结果均优于CPM算法和Core-Attachment算法.实验结果表明本算法在功能富集性方面比其他两种算法有一定的优越性.
4 小结
本文提出了一种新型的蛋白质复合物挖掘算法,首先利用GO术语相似性对蛋白质网络进行加权处理,然后通过蛋白质节点迭代传播,每个蛋白质节点都会从邻居节点集合中选取信号最强的信号类型来更新自己,直到整个蛋白质网络趋于稳定,算法终止.在实验中,本算法通过与CPM算法(k=3)和Core-Attachment算法对照,本算法在功能富集性方面和蛋白质复合物的匹配程度方面都有提高,说明本算法在识别蛋白质复合物上具有比较好的效果.
[1]WILKINS M R,SANEHEZ J C,GOOLEY A A,et al.Progress with proteome projects:why all proteins expressed by a genome should be identified and howtodoit[J].Biotechnologyand Genetic EngineeringReviews,1996,13(1):19-50.
[2]GARRELS J I.Yeast genomic databases and the challenge of the post-genomic era[J].Functional&Integrative Genomics,2002, 2(4):212-237.
[3]BARABASI AL,ALBERTR.Emergence ofscalingin randomnetworks[J].Seience,1992,86(5439):509-512.
[4]赵静,俞鸿,骆建华,等.应用复杂网络理论研究代谢网络的进展[J].科学通报,2006,51(11):1241-1245.
[5]LIN D.An information-theoretic definition of similarity[J].Proceedings of the Fifteenth International Conference on Machine Learning,1998,1(2):296-304.
[6]KROGAN N,CAGNEY G,YU H,et al.Global landscape of protein complexes in the yeast Saccharomyces cerevisiae[J].Nature, 2006,440(7084):637-643.
[7]PALLA G,DERENYI I,FARKAS I,et al.Uncovering the overlapping community structure of complex networks in nature and society[J].Nature,2005,435(7043):814-818.
[8]RZHETSKY A,GOMEZ S M.Birth of scale-free molecular networks and the number of distinct DNA and protein domains per genome[J].Bioinformatics,2001,17(10):988-996.
[9]PUS,WONGJ,TUMER B,et al.Up-to-date catalogues ofyeast protein complexes[J].Nucleic Acids Res,2009,37(3):825-831.
(责任编辑:卢奇)
Novel algorithm for mining of protein complexes
XU Rui,JIN Songlin,WANG Tingyu
(School ofInformation Engineering,Henan Institute ofScience and Technology,Xinxiang453003,China)
With the completion of the Human Genome Project,through analysis of structure characteristic of protein networks,identifying protein complexes or function modules is becoming the focus of current proteomics research.A novel algorithm for the mining of protein complexes was proposed.Based on an in-depth analysis of structure characteristics of protein networks,initially the protein network was weighted by GO terms of protein nodes, furthermore each protein node was divided into a stable protein set through the constant iteration of the nodes,finally protein complexes was identified in protein network.Compared with the algorithm CPM(k=3)and algorithm Core-Attachment,our method has a better performance in matching degree and functional enrichment of predicted protein complexes.
protein network;GO terms;protein interaction;protein complex
TP301.6
A
1008-7516(2017)01-0060-05
10.3969/j.issn.1008-7516.2017.01.012
2016-10-08
许睿(1987―),男,河南新乡人,硕士,助教.主要从事数据挖掘和生物信息学研究.
