H7N9
2017-04-11
H7N9
·编者按·
H7N9(influenza A virus subtype H7N9)属甲型流感病毒,是禽流感病毒的一个亚型。2013年3月,我国上海和安徽两地首先确诊H7N9禽流感病毒感染人的病例。此后,中国大陆其他地区、中国香港、中国台湾以及东南亚部分国家也发现了H7N9禽流感病毒感染人的病例,引起全球关注。国内外生物学家分析发现新型H7N9禽流感病毒起源于候鸟和家禽的多个基因重配,其中HA基因与华东地区家鸭群的H7N3病毒遗传关系最近,NA基因与2010/11香港地区的迁徙候鸟中分离到的(H11N9和H2N9)基因遗传关系最近,另外6个内部基因均来源于中国家禽中H9N2。H7亚型与当地不同宿主的H9N2病毒经过多次重组产生了新型H9N2,从水禽(家鸭)传递到陆生家禽(鸡、鹌鹑、鸽子等)体内。
截至目前,H7N9禽流感病毒感染人的病例仍以散发形式存在。目前已有H7N9禽流感病毒灭活疫苗、减毒活疫苗、重组疫苗完成了一期或二期临床研究,但距离疫苗获批使用尚有一段距离。
本专题得到赵卫教授(南方医科大学)的大力支持。
·热点数据排行·
截至2017年2月14日,中国知网(CNKI)和Web of Science(WOS)的数据报告显示,以“H7N9”为词条可以检索到的期刊文献分别为1446、941条,本专题将相关数据按照:研究机构发文数、作者发文数、期刊发文数、被引用频次进行排行,结果如下。

研究机构发文数量排名(CNKI)
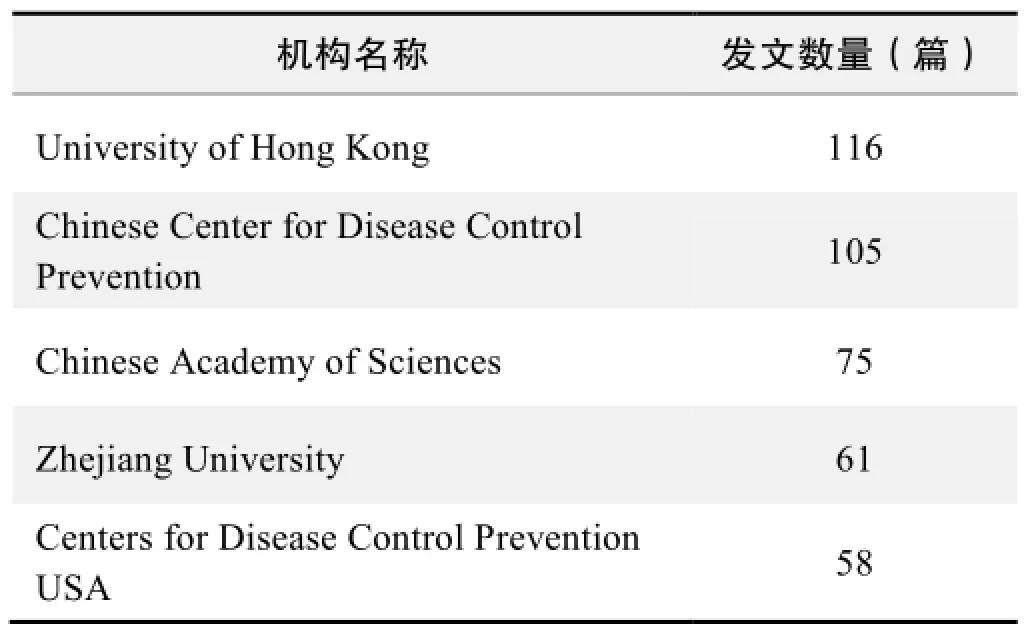
研究机构发文数量排名(WOS)

作者发文数量排名(CNKI)

作者发文数量排名(WOS)

作者发文数量排名(CNKI)(续表)
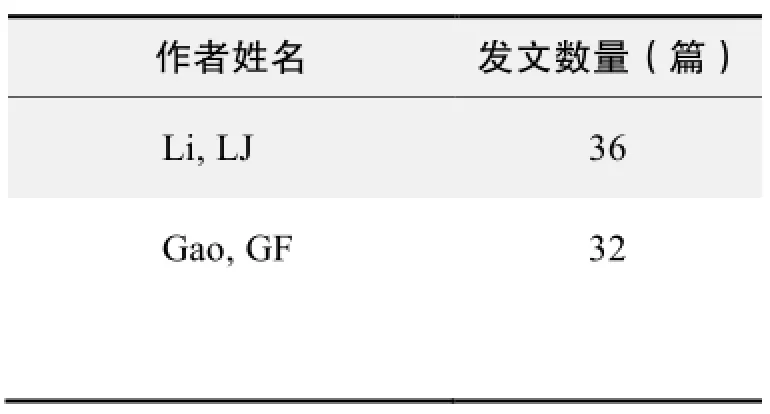
作者发文数量排名(WOS)(续表)
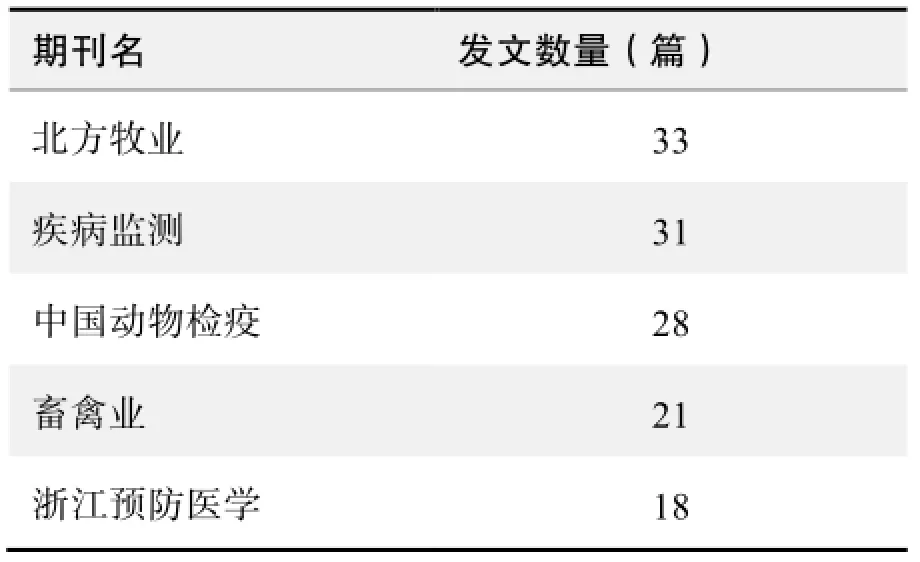
期刊发文数量排名(CNKI)

期刊发文数量排名(WOS)
(数据来源:中国知网、Web of Science,检索时间:2017-02-14)
根据中国知网(CNKI)数据报告,以“H7N9”等为词条可以检索到的高被引论文排行结果如下。
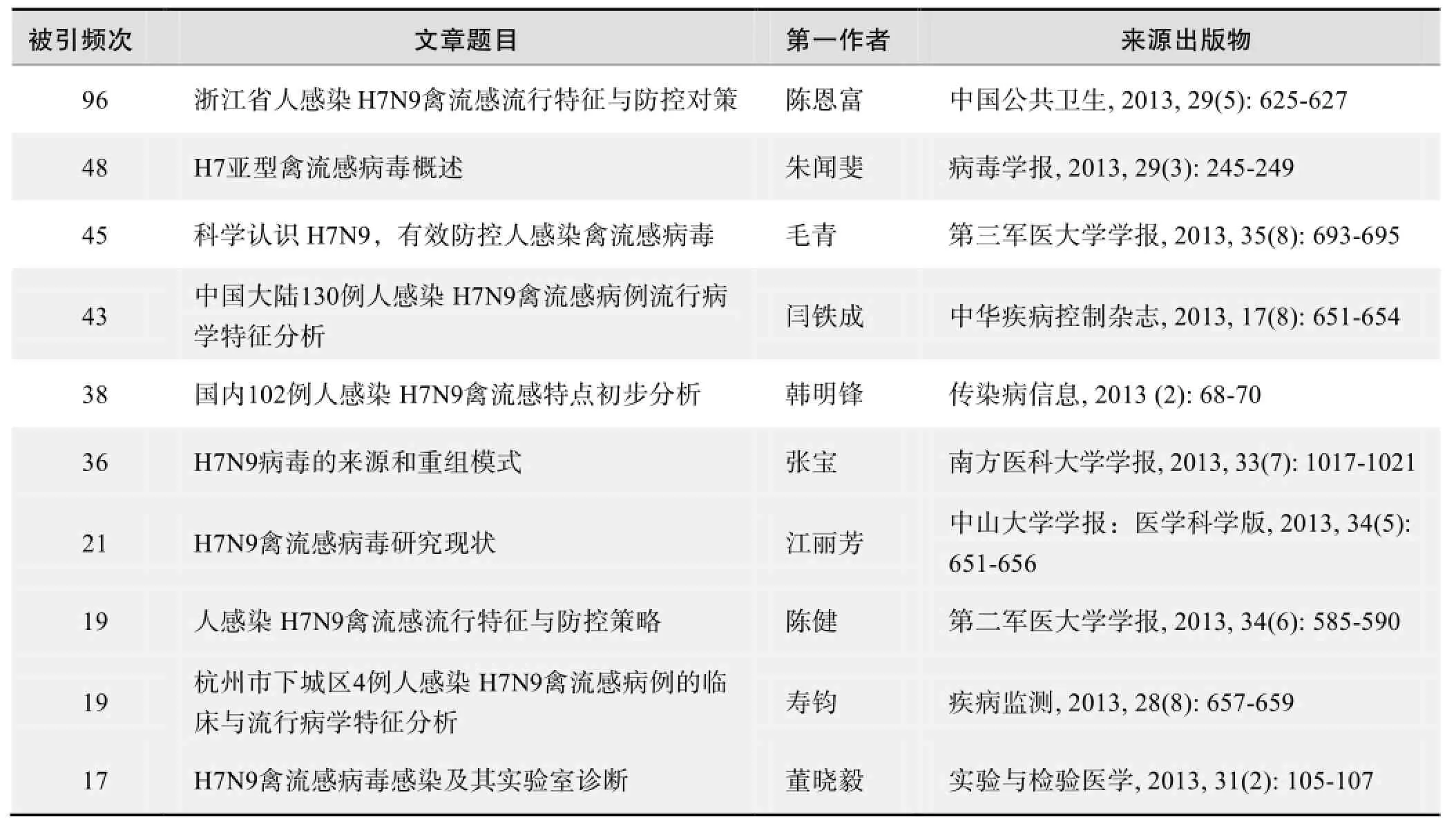
国内数据库高被引论文排行
根据Web of Science统计数据,以“H7N9”为词条可以检索到的高被引论文排行结果如下。

国外数据库高被引论文排行
·经典文献推荐·
基于Web of Science检索结果,利用Histcite软件选取LCS(Local Citation Score,本地引用次数)TOP 50文献作为节点进行分析,得到本领域推荐的经典文献如下。

本领域经典文献
Human infection with a novel avian-origin influenza A (H7N9) virus
Gao, RB; Cao, B; Hu, YW; et al.
Background: Infection of poultry with influenza A subtype H7 viruses occurs worldwide, but the introduction of this subtype to humans in Asia has not been observed previously. In March 2013, three urban residents of Shanghai or Anhui, China, presented with rapidly progressing lower respiratory tract infections and were found to be infected with a novel reassortant avianorigin influenza A (H7N9) virus. Methods: We obtained and analyzed clinical, epidemiologic, and virologic data from these patients. Respiratory specimens were tested for influenza and other respiratory viruses by means of real-time reverse-transcriptase-polymerase-chain-reaction assays, viral culturing, and sequence analyses. Results: A novel reassortant avian-origin influenza A (H7N9) virus was isolated from respiratory specimens obtained from all three patients and was identified as H7N9. Sequencing analyses revealed that all the genes from these three viruses were of avian origin, with six internal genes from avian influenza A (H9N2) viruses. Substitution Q226L (H3 numbering) at the 210-loop in the hemagglutinin
(HA) gene was found in the A/Anhui/1/2013 and A/Shanghai/2/2013 virus but not in the A/Shanghai/1/2013 virus. A T160A mutation was identified at the 150-loop in the HA gene of all three viruses. A deletion of five amino acids in the neuraminidase (NA) stalk region was found in all three viruses. All three patients presented with fever, cough, and dyspnea. Two of the patients had a history of recent exposure to poultry. Chest radiography revealed diffuse opacities and consolidation. Complications included acute respiratory distress syndrome and multiorgan failure. All three patients died. Conclusions: Novel reassortant H7N9 viruses were associated with severe and fatal respiratory disease in three patients.
来源出版物:New England Journal of Medicine, 2013, 368(20): 1888-1897
Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: Phylogenetic, structural, and coalescent analyses
Liu, D; Shi, WF; Shi, Y; et al.
Abstract: Background: On March 30, 2013, a novel avian influenza A H7N9 virus that infects human beings was identified. This virus had been detected in six provinces and municipal cities in China as of April 18, 2013. We correlated genomic sequences from avian influenza viruses with ecological information and did phylogeneticand coalescent analyses to extrapolate the potential origins of the virus and possible routes of reassortment events. Methods: We downloaded H7N9 virus genome sequences from the Global Initiative on Sharing Avian Influenza Data (GISAID) database and public sequences used from the Influenza Virus Resource. We constructed phylogenetic trees and did 1000 bootstrap replicates for each tree. Two rounds of phylogenetic analyses were done. We used at least 100 closely related sequences for each gene to infer the overall topology, removed suspicious sequences from the trees, and focused on the closest clades to the novel H7N9 viruses. We compared our tree topologies with those from a bayesian evolutionary analysis by sampling trees (BEAST) analysis. We used the bayesian Markov chain Monte Carlo method to jointly estimate phylogenies, divergence times, and other evolutionary parameters for all eight gene fragments. We used sequence alignment and homology-modelling methods to study specific mutations regarding phenotypes, specifically addressing the human receptor binding properties. Findings: The novel avian influenza A H7N9 virus originated from multiple reassortment events. The HA gene might have originated from avian influenza viruses of duck origin, and the NA gene might have transferred from migratory birds infected with avian influenza viruses along the east Asian flyway. The six internal genes of this virus probably originated from two different groups of H9N2 avian influenza viruses, which were isolated from chickens. Detailed analyses also showed that ducks and chickens probably acted as the intermediate hosts leading to the emergence of this virulent H7N9 virus. Genotypic and potential phenotypic differences imply that the isolates causing this outbreak form two separate subclades. Interpretation: The novel avian influenza A H7N9 virus might have evolved from at least four origins. Diversity among isolates implies that the H7N9 virus has evolved into at least two different lineages. Unknown intermediate hosts involved might be implicated, extensive global surveillance is needed, and domestic- poultry-toperson transmission should be closely watched in the future.
来源出版物:The Lancet, 2013, 381(9881): 1926-1932
H7N9 influenza viruses are transmissible in ferrets by respiratory droplet
Zhang, QY; Shi, JZ; Deng, GH; et al.
Abstract: A newly emerged H7N9 virus has caused 132 human infections with 37 deaths in China since 18 February 2013. Control measures in H7N9 virus-positive live poultry markets have reduced the number of infections; however, the character of the virus, including its pandemic potential, remains largely unknown. We systematically analyzed H7N9 viruses isolated from birds and humans. The viruses were genetically closely related and bound to human airway receptors; some also maintained the ability to bind to avian airway receptors. The viruses isolated from birds were nonpathogenic in chickens, ducks, and mice; however, the viruses isolated from humans caused up to 30% body weight loss in mice. Most importantly, one virus isolated from humans was highly transmissible in ferrets by respiratory droplet. Our findings indicate nothing to reduce the concern that these viruses can transmit between humans.
来源出版物:Science, 2013, 341(6144): 410-414
Biological features of novel avian influenza A (H7N9) virus
Zhou, JF; Wang, DY; Gao, RB
Abstract: Human infection associated with a novel reassortant avian influenza H7N9 virus has recently been identified in China. A total of 132 confirmed cases and 39 deaths have been reported. Most patients presented with severe pneumonia and acute respiratory distress syndrome. Although the first epidemic has subsided, the presence of a natural reservoir and the disease severity highlight the need to evaluate its risk on human public health and to understand the possible pathogenesis mechanism. Here we show that the emerging H7N9 avian influenza virus poses a potentially high risk to humans. We discover that the H7N9 virus can bind to both avian-type (α2,3-linked sialic acid) and human-type (α2,6-linked sialic acid) receptors. It can invade epithelial cells in the human lower respiratory tract and type II pneumonocytes in alveoli, and replicated efficiently inex vivo lung and trachea explant culture and several mammalian cell lines. In acute serum samples of H7N9-infected patients, increased levels of the chemokines and cytokines IP-10, MIG, MIP-1β, MCP-1, IL-6, IL-8 and IFN-α were detected. We note that the human population is naive to the H7N9 virus, and current seasonal vaccination could not provide protection.
来源出版物:Nature, 2013, 499(7459): 500-503
The genesis and source of the H7N9 influenza viruses causing human infections in China
Lam, TTY; Wang, J; Shen, YY; et al.
Abstract: A novel H7N9 influenza A virus first detected in March 2013 has since caused more than 130 human infections in China, resulting in 40 deaths(1,2). Preliminary analyses suggest that the virus is a reassortant of H7, N9 and H9N2 avian influenza viruses, and carries some amino acids associated with mammalian receptor binding, raising concerns of a new pandemic(1,3,4). However, neither the source populations of the H7N9 outbreak lineage nor the conditions for its genesis are fully known(5). Using a combination of active surveillance, screening of virus archives, and evolutionary analyses, here we show that H7 viruses probably transferred from domestic duck to chicken populations in China on at least two independent occasions. We show that the H7 viruses subsequently reassorted with enzootic H9N2 viruses to generate the H7N9 outbreak lineage, and a related previously unrecognized H7N7 lineage. The H7N9 outbreak lineage has spread over a large geographic region and is prevalent in chickens at live poultry markets, which are thought to be the immediate source of human infections. Whether the H7N9 outbreak lineage has, or will, become enzootic in China and neighbouring regions requires further investigation. The discovery here of a related H7N7 influenza virus in chickens that has the ability to infect mammals experimentally, suggests that H7 viruses may pose threats beyond the current outbreak. The continuing prevalence of H7 viruses in poultry could lead to the generation of highly pathogenic variants and further sporadic human infections, with a continued risk of the virus acquiring human-to-human transmissibility.
来源出版物:Nature, 2013, 502(7470): 241-244
