高效液相色谱法测定左旋四氢巴马汀自微乳中左旋四氢巴马汀含量
2017-04-08梁淑贞丁少波钟李青何瑞荣
梁淑贞,丁少波,钟李青,何瑞荣
(广东省东莞市人民医院药学部,广东东莞 523000)
高效液相色谱法测定左旋四氢巴马汀自微乳中左旋四氢巴马汀含量
梁淑贞,丁少波,钟李青,何瑞荣
(广东省东莞市人民医院药学部,广东东莞 523000)
目的建立测定左旋四氢巴马汀自微乳含量的高效液相色谱法。方法色谱柱采用Phenomenex Desc Gemini-NX 5 C18柱(250 mm×4.6 mm,5 m,110Å),流动相为0.1%磷酸溶液(三乙胺调pH至6.32)-甲醇(35∶65),检测波长为280 nm,进样量为10 L,流速为1.0 mL/min,柱温为30℃。结果左旋四氢巴马汀质量浓度在0.1~1.0 g/L范围内与峰面积线性关系良好(r=0.999 9);精密度试验RSD为0.38%;稳定性试验RSD为0.96%;重复性试验左旋四氢巴马汀的含量为91.81%,RSD为0.58%;回收试验平均回收率为99.97%,RSD为0.60%(n=9)。结论该方法简便、灵敏、重复性好,结果准确、可靠,可作为左旋四氢巴马汀自微乳的质量控制方法。
高效液相色谱法;左旋四氢巴马汀自微乳;含量测定
左旋四氢巴马汀是常用的镇静、止痛药,其还对心肌损伤、脑损伤有保护作用,具有抗心律失常的功效。也有研究表明,左旋四氢巴马汀能提高海洛因成瘾者的戒毒率[1-2]。但由于其水溶性极差,口服生物利用度极低,故其口服制剂应用受到限制[3]。自微乳化药物传递系统(self-emulsifying drug delivery system,SEDDS)是一种新型药物传递系统,药物溶解于载体中,口服给药后在胃肠道的蠕动下自发形成O/W型微剂,粒径10~100 nm。SEDDS能提高难溶性药物的溶解度,增加细胞膜的流动性,促进药物淋巴吸收,从而提高药物的生物利用度[4]。为了控制左旋四氢巴马汀自微乳的质量,本研究中建立了左旋四氢巴马汀自微乳的含量测定方法,现报道如下。
1 仪器与试药
1.1 仪器
LC-20A型高效液相色谱仪(日本岛津公司); SHB-Ⅲ型循环水式真空泵(巩义市予华仪器有限公司);SHA-CA型数显水浴恒温振荡器(金坛市易辰仪器制造有限公司);FA2004型电子分析天平;TDL-5A型高速离心机(上海菲恰尔分析仪器有限公司);KQ-500DB型数控超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药
左旋四氢巴马汀对照品(中国药品生物制品检定所,批号为100452-200301);左旋四氢巴马汀原料药(西安开来生物工程有限公司,纯度≥98%,批号为K120105,K120106);聚乙二醇400(PEG400,西陇化工股份有限公司,批号为130115);甲醇为色谱纯,其余试剂均为分析纯。
2 方法与结果
2.1 自微乳制备
称取左旋四氢巴马汀原料药200 mg(主药),乙酸异丙酯10 g(辅料),OP乳化剂10 g(辅料),PEG400 1 g (辅料),精密称定,置具塞锥形瓶中,于(37±0.5)℃恒温水浴振荡至原料药完全溶解,制成自微乳,称重。平行制备3批左旋四氢巴马汀自微乳。
2.2 含量测定
2.2.1 色谱条件[5-16]
色谱柱:Phenomenex Desc Gemini-NX 5 μ C18柱(250 mm×4.6 mm,5 μm,100Å);流动相:0.1%磷酸溶液(三乙胺调pH至6.32)-甲醇(35∶65);检测波长:280 nm;进样量:10 μL;流速:1.0 mL/min;柱温:30℃。
2.2.2 溶液制备
对照品溶液:取左旋四氢巴马汀对照品50 mg,精密称定,置50 mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,制成质量浓度为1 g/L的对照品贮备液。
供试品溶液:取2.1项下左旋四氢巴马汀自微乳2 g,精密称定,置50 mL容量瓶中,用流动相溶解并稀释至刻度,摇匀,即得。
空白自微乳溶液:取2.1项下左旋四氢巴马汀自微乳处方量辅料,置具塞锥形瓶,于37℃恒温水浴振荡2 h,取2 g,精密称定,置50 mL容量瓶中,用流动相溶解并稀释至刻度,摇匀,即得。
2.2.3 方法学考察
系统适用性试验:取2.2.2项下3种溶液,按拟订色谱条件分别进样测定,色谱图见图1。结果表明,左旋四氢巴马汀自微乳对照品溶液与供试品溶液的保留时间一致,空白自微乳溶液在相应位置无干扰峰出现,同时主峰与其他组分分离完全,拖尾因子在0.95~1.05,分离度大于1.5,理论板数计算不低于3 000。
线性关系考察:精密吸取对照品溶液1.0,2.0,4.0,6.0,8.0,10.0 mL,分别置10 mL容量瓶中,加甲醇稀释至刻度,摇匀,制成质量浓度分别为0.1,0.2,0.4,0.6,0.8,1.0 g/L的系列对照品溶液。按拟订色谱条件进样测定,以进样质量浓度(X,g/L)为横坐标、峰面积(Y)为纵坐标绘制标准曲线,得回归方程Y=436.58X+0.947 2,r=0.999 9(n=6)。结果表明,左旋四氢巴马汀质量浓度在0.1~1.0 g/L范围内与峰面积线性关系良好。
精密度试验:取线性关系考察项下质量浓度为0.4 g/L的对照品溶液,按拟订色谱条件连续进样6次,记录峰面积。结果峰面积分别为176.7,177.0,176.5,176.1,176.9,178.1,平均176.9,RSD为0.38%(n=6),表明仪器精密度良好。
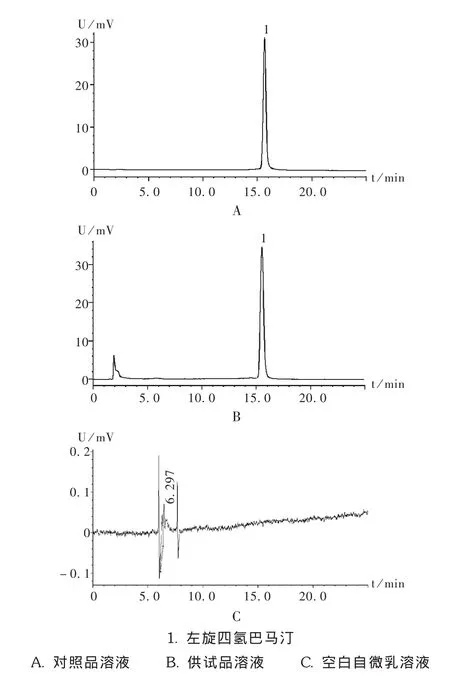
图1 高效液相色谱图
重复性试验:取同一批(批号为160404)左旋四氢巴马汀自微乳,按供试品溶液制备方法制备溶液,按拟订色谱条件连续进样测定。结果左旋四氢巴马汀的含量分别为90.97%,91.92%,92.56%,92.11%,91.64%,91.74%,平均91.81%,RSD为0.58%(n=6),表明方法重复性良好。
稳定性试验:取同一供试品溶液于室温放置,分别于0,2,4,6,8,10,12 h时按拟订色谱条件进样测定。结果左旋四氢巴马汀峰面积分别为164.8,166.9,165.9,167.2,168.8,168.9,平均167.1,RSD为0.96%(n=6),表明供试品溶液在室温下12 h内稳定。
加样回收试验:取同一批(批号为160404)左旋四氢巴马汀自微乳1 g,共9份,精密称定,置50 mL容量瓶中,并分别精密加入左旋四氢巴马汀对照品6,8,10 mg,按拟订色谱条件进样测定,计算回收率。结果见表1。
2.2.4 样品含量测定
取3批(批号为160404,160405,160406)左旋四氢巴马汀自微乳,依法制备供试品溶液,进样测定,计算含量。结果见表2。
3 讨论
3.1 流动相选择
左旋四氢巴马汀不溶于水,可溶于甲醇,故用含65%甲醇的流动相溶解,流动相中加入三乙胺可改善主峰的拖尾现象。该制剂是O/W型乳剂,在流动相中溶解性能很好,且不影响左旋四氢巴马汀的含量测定。曾考察甲醇-磷酸盐溶液、甲醇-水溶液、甲醇-磷酸溶液、乙腈-磷酸溶液,有机相占60%以上,大部分用了三乙胺等扫尾剂。甲醇与水的流动相组合预试验发现,峰形不佳;甲醇-磷酸盐流动相系统峰形较好,样品测定也未见有干扰,但流动相对于样品的溶解效果不是很好,需要超声处理;甲醇-磷酸溶液流动相系统未加三乙胺时峰形不好,考虑加入扫尾剂。采用单因素对照试验优选加入三乙胺的量,最终流动相pH为6.3时,峰形最佳,且流动相对供试品的溶解较好。
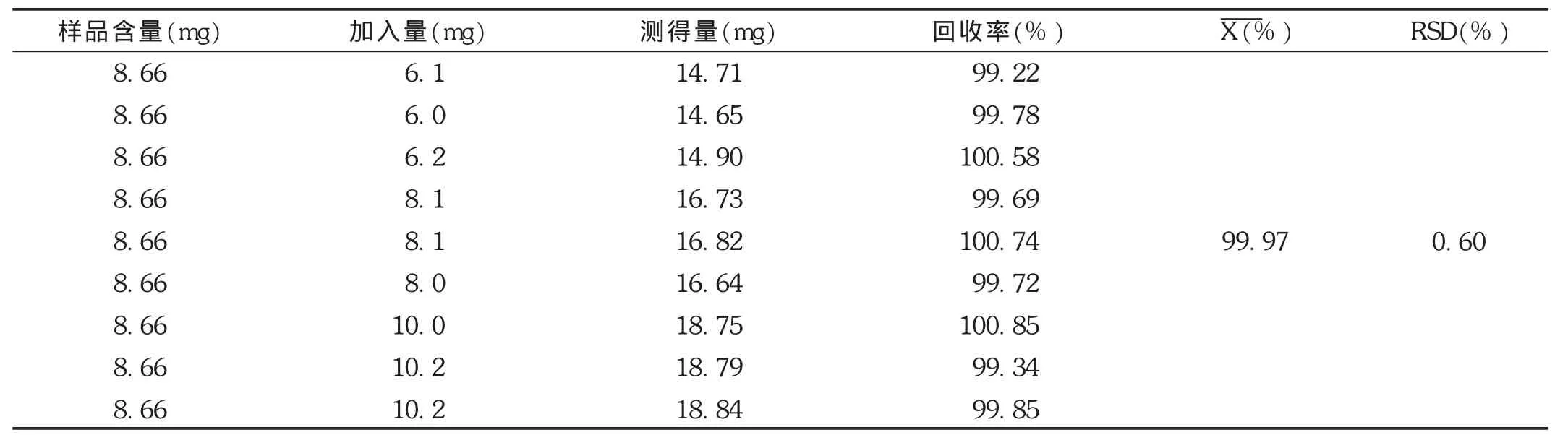
表1 左旋四氢巴马汀加样回收试验结果(n=9)

表2 样品含量测定结果(n=3,%)
3.2 处方筛选
通过溶解度试验筛选油相、乳化剂和助乳化剂,并以乳化时间、色泽和乳化颗粒形态为指标,通过伪三相图的绘制,筛选最佳处方及配比,制备空白自微乳及左旋四氢巴马汀自微乳。
3.3 波长选择
有文献显示,通过全波长扫描试验,结果281 nm波长处有最大吸收峰。故选择280 nm作为检测波长,且无干扰峰出现,方法学考察结果较好。
左旋四氢巴马汀属生物碱类,难溶于水,口服生物利用度较低,受光热极易氧化为巴马汀,故含量测定时对样品的处理不能采用加热溶解的方式。高效液相色谱法测定含量快速、准确,样品的处理经过溶解即可。方法学考察各项指标符合规定,精密度、稳定性、准确度都符合要求,可作为乳剂生产过程的质量控制和检测方法,也可作为自微乳成品的质量标准。
[1]徐婷,金昔陆,曹惠明.延胡索乙素药理作用的研究进展[J].中国临床药学杂志,2001,10(1):58-60.
[2]王燕波,任燕华,郑继旺,等.左旋四氢巴马汀对吗啡条件性位置偏爱的影响[J].中国药理学通报,2005,21(12):1442-1445.
[3]萧伟斌.左旋四氢巴马汀体内外代谢处置研究[D].北京:中国人民解放军军事医学科学院,2016.
[4]沈海蓉,李中东,钟明康.自微乳释药系统及其制剂的研究进展[J].中国新药与临床杂志,2005,24(5):409-412.
[5]罗肖雪,陈光英,张一,等.反相HPLC法测定小叶地不容中四氢巴马汀的含量[J].中药材,2006,29(10):1050-1051.
[6]池秀珍.高效液相色谱法测定复方枣仁胶囊中左旋延胡索乙素含量[J].中国药业,2009,18(10):40-42.
[7]马小亚,梁颖彬,邢剑锋,等.反相高效液相色谱法测定血浆中左旋四氢巴马汀浓度[J].药物分析杂志,1994,14(2): 13-15.
[8]黄兴振,龙翊婷,蒋伟哲,等.左旋四氢巴马汀分散片的鉴别及含量测定[J].华西药学杂志,2014,29(3):334-336.
[9]黎远冬,蒋湘.高效液相色谱法测定复方金蒲片中左旋四氢帕马丁的含量[J].药物分析杂志,2005,25(9):1139-1140.
[10]陈旭,陆兔林,张先洪.HPLC测定延胡索不同炮制品中延胡索乙素含量[J].中成药,2003,25(9):38-39.
[11]窦志英,孙巍.不同炮制方法对延胡索中延胡索乙素含量的影响[J].天津中医药大学学报,2006,25(3):186-187.
[12]张捷,黄荣华.HPLC测定延胡索不同炮制品中延胡索乙素含量[J].西北药学杂志,2006,21(5):209-210.
[13]刘迎春,牛晚扬,段丽颖,等.RP-HPLC法测定不同产地延胡索中延胡索乙素含量[J].化学试剂,2007,29(2):97-98.
[14]袁双容,林涛,谢晓密.延胡索中延胡索乙素含量测定方法的探讨[J].广州医药,2007,38(5):50-52.
[15]龚青,周蒂,王碧娟.HPLC法测定延胡索中延胡索乙素的含量[J].中国现代应用药学,2000,17(4):315-317.
[16]韩建伟,张莉,高小春.HPLC法测定复方元胡止痛膏贴中延胡索乙素含量[J].中国药师,2009,12(5):621-623.
Content Determination of L-Tetrahydropalmatine in L-Tetrahydaopalmatine Self-Microemulsion by HPLC
Liang Shuzhen,Ding Shaobo,Zhong Liqing,He Ruirong
(Department of Pharmacy,Dongguan People′s Hospital,Dongguan,Guangdong,China523000)
Objective To establish a content determination method of L-tetrahydropalmatine in L-tetrahydaopalmatine Self-Microemulsion by HPLC.MethodsThe column was Phenomenex Desc Gemini-NX 5 μ C18column(250 mm×4.6 mm,5 μm,110Å);the mobile phase consisted of 0.1%phosphoric acid solution(pH adjusted to 6.32 with triethylamine)and methanol(35∶65);the detection wavelength was set at 280 nm;the injection volume was 10 μL;the flow rate was 1.0 mL/min and the column temperature was 30℃.ResultsThe calibration curve was linear in the range of 0.1 to 1.0 g/L(r=0.999 9);RSD of accuracy was 0.38%,and the RSD of stability was 0.96%.The content of L-tetrahydropalmatine was 91.81%and the RSD was 0.58%in the repeatability test.The recovery rate of L-tetrahydropalmatine was 99.97%and the RSD was 0.60%(n=9)in the recovery test.ConclusionThe method is simple,sensitive,reproducible,accurate and reliable,which can be used as a quality control method for L-tetrahydropalmatine self-microemulsion.
HPLC;L-tetrahydropalmatine Self-Microemulsion;content determination
R927.2;R971+.3
A
1006-4931(2017)01-0028-03
10.3969/j.issn.1006-4931.2017.01.009
2016-10-01;
2016-11-21)
梁淑贞,大学本科,主管药师,主要从事医院药学工作,(电子信箱)120714659@qq.com。
