As掺杂浓度对FeS2电子结构及光学性质的影响
2017-04-06马万坤陈建华张国范李玉琼冯其明
马万坤,陈建华,张国范,李玉琼,冯其明
As掺杂浓度对FeS2电子结构及光学性质的影响
马万坤1,陈建华2,张国范1,李玉琼2,冯其明1
(1. 中南大学资源加工与生物工程学院,长沙 410083;2. 广西大学资源与冶金学院,南宁 530004)
采用基于密度泛函理论框架下的第一性原理平面波超软赝势法,对不同As掺杂浓度FeS2的几何结构、电子结构和光学性质进行计算和讨论。采用2×2×2(Fe32S63As),2×2×1(Fe16S31As)和2×1×1(Fe8S15As)的超晶胞模型,用1个As原子取代1个S原子,使掺杂浓度分别为1.93%、3.82%和7.48%(质量分数),然后进行3种掺杂体系的计算,并将其与理想体系进行对比。几何结构与电子结构的计算结果表明:As掺杂使得FeS2的晶格常数和晶胞体积增大,导带部分下移,禁带宽度减小,并且在−10.4~−9.5 eV的浅部价带产生了由As的p态贡献的杂质能级。光学性质计算结果表明,掺杂后Fe(S1−xAs)2的静态介电常数、折射率和光电导率在一定范围内均随着掺杂量的增大而明显增大,说明As掺杂显著增强了FeS2对光的吸收以及光电转换效率。
As掺杂;黄铁矿;光电性质;第一性原理
随着能源需求的激增和环境污染的加剧,太阳能电池已成为世界各国研究的热点。硅基太阳能电池虽然具有能量转化效率高和稳定性好等优点,但高昂的制造成本限制了其大规模应用[1−3]。因而,继续研究并开发其他新型的太阳能电池材料及先进的太阳能电池系统,是目前极为重要的课题。金属硫化物因其半导体属性在太阳能电池、热电材料以及存储装置等领域显示出了广阔的应用前景,因而得到了广泛关注。通过切实可行的途径使金属硫化物具备优良的光学、电学或者磁学性能是其能够得到有效应用的先决条件。
黄铁矿(FeS2)是地壳中最常见的硫化矿,组成元素无毒且地球储量丰富。立方晶系黄铁矿具有优异的半导体特性:合适的禁带宽度(g≈0.95 eV)和较高的光吸收系数(≈5×105/cm,≤700 nm)[4−5],其环境相容性好,制备成本低廉,是一种较有研究价值的新型太阳能电池材料[6−12]。已有研究采用人工合成方法制备出FeS2薄膜,尽管其易于氧化降解,但仍表现出良好的光电性能[13]。1986年,CHATZITHEODOROU等[14]首次制备出FeS2光电化学电池和肖特基太阳能电池,但是较低的光电压限制了FeS2在光伏器件方面的应用。
天然黄铁矿物中存在不同类型的杂质和含量,黄铁矿的光电催化性质会受到这些杂质的影响。LEHNER等[15]使用化学气相沉积来制备Ni掺杂的黄铁矿薄膜,证明Ni的掺入在光电导性测量中会显著影响光电导的敏感性。FERRER等[16]制备了掺Cu黄铁矿薄膜,掺杂后电阻率从0.002 Ω·cm增加到3.28 Ω·cm,掺杂体系光吸收边从0.8 eV增大到1.1 eV。YU等[17]制备了掺Mn的黄铁矿薄膜,掺杂后薄膜形貌从颗粒状变为片状,有利于光捕获。李玉琼等[18−21]研究了Co、Ni、As等20余种天然杂质对黄铁矿电子结构和光学性质的影响,结果表明:Co、Ni、As的掺杂均使得黄铁矿的吸收带边发生了明显的红移。尽管国内外研究人员对黄铁矿薄膜的合成制备、As掺杂纳米晶薄膜的研究已经很多,但对As掺杂对黄铁矿光学性质的影响目前仍鲜见报道。
本文作者以FeS2超晶胞作为基体,以As为掺杂原子,采用基于密度泛函理论的第一性原理平面波超软赝势法,对不同浓度As掺杂FeS2前后的能带、电子态密度和光学性质进行了对比和研究,初步揭示了 通过As掺杂FeS2改变其光电性质的内在原因,为开发FeS2光伏材料的实验工作提供了理论参考。
1 理论模型及计算方法
半导体化合物FeS2属于等轴晶系,其空间群为,每个晶胞含有4个FeS2分子单元,Fe原子分布在晶胞的6个面心和8个顶角上,每个Fe原子与6个相邻的硫原子配位,每个S原子与3个Fe原子和1个S原子配位,2个硫原子之间形成哑铃状结构,以硫二聚体形式存在,且沿着(111)方向排列。FeS2的晶格常数为===0.5379 nm,===90°。
在本计算中,首先采用单胞模型描述理想FeS2的电子结构及性质,然后分别采用2×2×2 (Fe32S63As)、2×2×1(Fe16S31As)和2×1×1(Fe8S15As) 3个超晶胞模型,用1个As原子取代1个S原子,使掺杂浓度分别为1.93%、3.82%和7.48%(质量分数),然后进行3种掺杂体系的计算。通过计算得到3种掺杂浓度下FeS2的优化结构、掺杂前后的能带结构、电子态密度及光学性质。计算模型如图1所示。
本计算采用基于密度泛函理论(DFT)框架下的第一性原理赝势平面波法,主要的计算工作由CASTEP软件包[22]完成。计算中采用超软赝势(USPP)[23]来处理离子实与电子之间的相互作用,交换关联泛函采用广义梯度近似(GGA)下的PW91梯度修正近似。平面波截断能测试表明,截断能取360 eV较为合理。计算选取的价电子为:Fe 3d64s2、S 3s23p4和As 4s24p3。采用BFGS算法对体系进行几何优化,得到稳定的结构,优化的收敛标准为:原子位移的收敛阈值为0.0002 nm,原子间作用力的收敛阈值为0.8 eV/nm,原子间的内应力收敛阈值为0.1 GPa,最大能量改变的收敛阈值为2.0×10−5eV/atom,自洽计算收敛精度设置为每个原子1 meV,布里渊区的积分分别采用3×5×3、3×3×2和2×2×1的MONKHORST和PACK[24]特殊点对全Brillouin区求和。
2 结果与讨论
2.1 几何结构优化结果
未掺杂的FeS2单胞优化后及As掺杂FeS2后的超晶胞模型经几何优化后折合的晶胞参数、总能量的晶格常数值如表1所列。从表1可以看出,优化后FeS2的晶格参数与实验值(5.416 nm)[25]非常接近,平衡晶格常数比实验值小0.68%。随着As掺杂浓度增大,Fe(S1−xAs)2(=0.0156, 0.0312, 0.0625)的晶格常数和晶格体积均逐渐增大。引起这种变化的主要原因是As的共价半径为0.120 nm,而S的共价半径为0.102 nm,As的共价半径比S的大,当用As来置换S时,就会引起晶胞体积增大。晶胞优化的结果与文献[26]的结果一致。
图1 FeS2单胞及As掺杂FeS2的超晶胞结构图

表1 结构优化后Fe(S1−xAsx)2 (x=0, 0.0156, 0.03125, 0.0625)的折合晶胞参数和总能
Total energy: Total energy of Fe(S1−xAs)2convert to energy in 2×2×2 super cell.
2.2 电子结构
2.2.1 电子态密度
图2所示为FeS2单胞结构优化后的总态密度及Fe、S各亚层电子的分波态密度图。在FeS2晶体结构中,处于八面体中心的铁原子由于晶体场作用Fe 3d轨道分裂成2g和g*两部分,因此Fe 3d轨道间断地分布在价带和导带中。Fe 3d的2g部分主要出现在上部价带(靠近费米能级),与S 3p态重叠较小,表明它们之间的成键作用较弱。由图2(b)可知,Fe的3d2g没有实质上的成键−反键劈裂,其本质原因是轨道的对称性和能量不匹配,因而其也被称作非键轨道[27]。 Fe 3dg*和S 3p形成反键,反键峰约在2.5 eV处, Fe 3dg*与S 3p态密度重叠程度较高,表明这二者之间作用较强。
在态密度图中,轨道的贡献就意味着轨道重叠,重叠度越大,轨道杂化就越强烈。从图2(b)中可以看出,Fe的3d、4s轨道以及S的3s、3p轨道在−3.2 eV的同一能量位置出现了态密度峰,说明这两个轨道发生了重叠形成了一个新的轨道,从杂化峰的宽度来看Fe、S杂化峰离域性强,轨道杂化作用较强。从图2可以看出,在能量为−17.5~−9 eV范围内,态密度几乎全部由S 3s态贡献,仅有少量的Fe 4s和S 3p态贡献;在能量为−7~−1.5 eV范围内,态密度主要由Fe 3d态和S 3p态贡献,其中S 3p态贡献最大,而Fe 4s和S 3s态的贡献相对较小;费米能级附近价带区的态密度,则主要由S的3p态和非成键Fe 3d态组成,且大部分由Fe 3d态贡献。费米能级附近导带区0~4 eV范围内的态密度主要由S 3p和Fe的反键3dg*态贡献,而S 3s态贡献很小。
从图2(b)中还可以看出,S 3s态对深部价带的贡献较大,而S 3p态则主要对浅部价带产生贡献。Fe 4s对整个能带范围态密度的贡献相对较少,而费米能级附近的态密度主要来自Fe 3d的贡献,这和本征态FeS2的总态密度计算结果相符合。

图2 FeS2单胞总态密度和各原子态密度
As掺杂置换S后得到的Fe(S1−As)2态密度如图3所示。从图3中可以看出,As置换FeS2中的S原子后,由于As和S原子核外电子排布的相似性,对FeS2的电子态密度影响不是很大,价带顶和导带底仍主要由Fe 3d态决定,As的s层和p层电子分别与S的s层和p层电子混合,共同构成Fe(S1−As)2的能态密度。
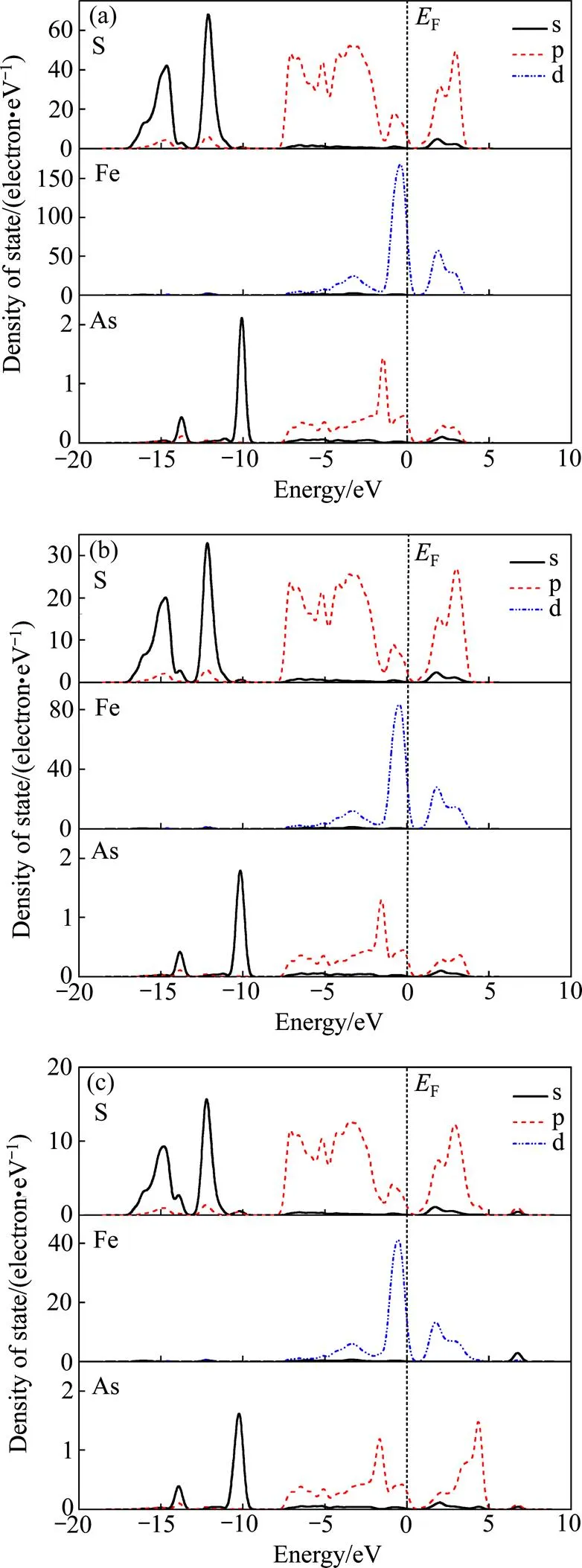
图3 Fe(S1−xAsx)2的态密度图
As的掺杂使FeS2的电子能带在浅部价带产生了由它的p态贡献的杂质能级,并在更低的能量范围内产生了由其s态贡献的杂质能级,并且随着体系As掺杂浓度的增大,这种贡献越来越明显。
2.2.2 能带结构
图4所示为As掺杂前后FeS2和Fe(S1−As)2的能带结构,图中G、F、Q、Z、G为FeS2晶体第一布里渊区高对称点。为了进行对比,将理想FeS2的第一布里渊区沿对称方向的点取样设置为与掺杂后的体系一致。在计算中所考虑的禁带宽度是从价带最高点到导带最低点之间的距离,计算得到理想FeS2的禁带宽度为0.55 eV,小于实验值0.95 eV,一般认为出现这种结果的主要原因是作为基态理论的DFT,其Kohn-Sham方程的本征值不能给出系统的激发态能量,导致位于导带部分的电子态能量值比实验值小,从而带隙偏小,一般情况下能带值与理论值的误差范围为30%~50%[28]。
As掺杂后,FeS2的带隙为0.43 eV,这表明As掺杂后FeS2的禁带宽度变小。从图4(b)、(d)、(f)可以明显看出,价带的上方出现了明显的杂质能带,对比图4(e)、(f)可以看出,理想FeS2费米能级附近的导带带宽为0.55~4.10 eV,而掺杂As后费米能级附近的导带带宽变为0.45~4.41 eV,导带不仅变宽而且还下移了0.1 eV,也说明掺杂后FeS2的禁带宽度变小。
2.3 光学性质计算结果与讨论
为了研究As掺杂对FeS2的光学性质的影响,对Fe(S1−As)2的光学跃迁特性进行了研究。在线性响应范围内固体宏观光学响应函数通常可以由光的介电函数() =1() + i2() 或折射率() =() +i() 来描述,体系在较小波矢下对光场的线性响应由介电函数的虚部2()决定,它可以通过占据态和未占据态的动量跃迁矩阵元计算得到。
2.3.1 复介电函数
介电常数的虚部2()对任何材料来说都是非常重要的,它作为沟通带间跃迁微观物理过程与固体电子结构的桥梁,反映了固体能带结构及其他各种光谱信息。图5所示为计算得到的未掺杂FeS2和Fe(S1−xAs)2的介电函数实部和虚部。
由图5(a)可知,未掺杂的FeS2的静态介电常数1(0)=19.6,随着As掺杂量的增加,Fe(S1−xAs)2的静态介电常数1(0)的值逐渐增大。当<0.46 eV或>9.5 eV时Fe(S1−xAs)2的1值比未掺杂时的大,而当0.46 eV<<9.5 eV时,Fe(S1−xAs)2的1值比未掺杂的FeS2的小。在<7.5 eV的范围内,1值随着掺杂量的增加逐渐增大,而>7.5 eV后,Fe(S1−xAs)2的1值随掺杂量的增加而减小并且逐渐接近未掺杂FeS2的1值。

图4 FeS2和Fe(S1−xAsx)2 (x=0.0156, 0.0312, 0.0625) 对应的相同超胞的能带结构图

图5 FeS2和Fe(S1−xAsx)2的介电函数实部和虚部
由图5(b)可知,对于未掺杂的FeS2在<0.28 eV的能量范围内2=0,2()在图所示的能量范围内有3个明显的介电峰,对应的能量依次为2.63、7.26和9.48 eV。位于能量为2.63 eV的第一个介电峰来自S的s和p轨道发生杂化后电子到导带的跃迁,能量为7.26和9.48 eV的介电峰来源于价带中部S 3p态到导带Fe 3d态的电子跃迁。当As掺杂后,可以明显看到2()的第一跃迁峰向低能方向移动,并且第一跃迁峰的值随掺杂量的增加而明显增大。当<1.28 eV时,Fe(S1−xAs)2的光跃迁强度远大于未掺杂的FeS2,且光跃迁强度随掺杂量的增大而增大,说明As掺杂能够增强电子在<1.28 eV的低能量范围内的光学跃迁。在>2.5 eV的能量范围Fe(S1−xAs)2的光跃迁强度小于未掺杂的FeS2,且光跃迁强度随着As掺杂量的增加而增大。
2.3.2 复折射率
由公式1=2−2和2=2可以从复介电函数得到FeS2的复折射率。图6所示为FeS2和Fe(S1−xAs)2的折射率和消光系数。对于FeS2理想体系,折射率(见图6(a))在=1.66 eV处取得最大值,当>1.66 eV时,折射率快速下降,在6.3~9.4 eV范围内出现两个变化平缓的峰,表明FeS2在这一能量范围内呈现出一定的金属反射特性。消光系数(见图6(b))在3.88 eV处取得最大值,随后随着能量的增加而减小,并在能量约为20 eV时减小到零。
由图6(a)可知,当As掺杂量从本征态增加到=0.0625时,折射率的值从4.43增加到5.99。当0.56~14.1 eV时,Fe(S1-xAs)2的折射率小于FeS2的折射率,且折射率随掺杂量的增加而有小幅增大。在<0.56 eV和>14.1 eV的范围内,Fe(S1−xAs)2的折射率大于FeS2的折射率,且随掺杂量的增大而减小。消光系数(见图6(b))在<1.42 eV范围内出现了新的峰值,并且As掺杂后消光系数较本征态均有所增大。在3.48~10.54 eV的能量范围内出现了较强的波动峰,当>10.54 eV时,随掺杂量的增加逐渐增大。
2.3.3 光电导率
光电导率是光电子材料的一个重要参数,它描述了光照引起半导体电导率改变的现象。图7所示为Fe(S1−xAs)2和FeS2的光电导率实部1()。从图7可以看出,FeS2的光电导率与介电函数的虚部是对应的,1()呈先上升后下降的变化趋势,其中在=3.60 eV处达到第一个峰值,在=7.49和9.65 eV处分别达到第二、第三个峰值,且在=3.60 eV处取得最大值。当掺入As后,除Fe(S1−xAs)2(=0.0625)在4.8~5.5 eV范围内的光电导率大于本征态外,掺杂体系的光电导率均小于本征态FeS2的光电导率,同时,在>2.66 eV时随着掺杂量的增加,Fe(S1−xAs)2的光电导率逐渐增加。Fe(S1−xAs)2的光电导率最大峰值出现在2.9~3.3 eV的范围内,随着As掺杂浓度的增大,掺杂体系的电导率逐渐增大,峰值出现的位置也随掺杂量的增加向高能方向移动。

图7 FeS2和Fe(S1−xAsx)2的光电导率
3 结论
1) 对不同As掺杂浓度FeS2的几何结构计算结果表明,As掺杂FeS2引起了晶格参数的增大,随As掺杂浓度增大,Fe(S1−xAs)2(=0.0156,0.0312,0.0625)的晶格常数和晶格体积均逐渐增大,这是由于As的共价半径大于S的共价半径所致。
2) 理想FeS2的优化表明其禁带宽度为0.55 eV,费米能级附近导带区0~4 eV范围内的态密度主要由S 3p和Fe的反键3dg*态贡献,而S 3s态贡献很小。As掺杂使得FeS2导带下移,禁带宽度减小,并且在−10.4~−9.5 eV的浅部价带产生了由As的p态贡献的杂质能级。通过对单个原子态密度的分析发现,As的掺杂对黄铁矿费米能级处电子态密度有贡献,且对Fe和S原子的态密度分布产生了影响。
3) 对光学性质计算结果分析可知,Fe(S1−xAs)2的静态介电常数、第一介电峰、折射率随着As掺杂量的增大而逐渐增大,平均反射效应的减弱有效地提高了其光电转换效率。因此,可以通过对FeS2的As掺杂以及调整掺杂浓度来得到不同光电特性的FeS2电催化活性材料。
[1] SONG Xiao-hui, WANG Min-qiang, ZHANG Hao, DENG Jian-ping, YANG Zhi, RAN Chen-xin, YAO Xi. Morphologically controlled electrodeposition of CdSe on mesoporous TiO2film for quantum dot-sensitized solar cells[J]. Electrochimica Acta, 2013, 108(10): 449−457.
[2] SUEZAKI T, CHEN J I L, HATAYAMA T, FUYUKI T, OZIN G A. Electrical properties of p-type and n-type doped inverse silicon opals-towards optically amplified silicon solar cells[J]. Applied Physics Letter, 2010, 96(24): 242102-1−242102-3.
[3] ZHAI Guang-mei, BEZRYADINA A, BREEZE A J, ZHANG Dao-li, ALERS G B, CARTER S A. Air stability of TiO2/PbS colloidal nanoparticle solar cells and its impact on power efficiency[J]. Applied Physics Letter, 2011, 99(6): 063512-1−063512-3.
[4] ALTERMATT P P, KIESEWETTER T, ELLMER K, TRIBUTSCH H. Specifying targets of future research in photovoltaic devices containing pyrite (FeS2) by numerical modeling[J]. Solar Energy Materials and Solar Cells, 2002, 71(2): 181−195.
[5] MIGUEL C A, FABER M S, TAN Yi-zheng, HAMERS R J, JIN Song. Synthesis and properties of semiconducting iron pyrite (FeS2) nanowires[J]. Nano Letter, 2012, 12(4): 1977−1982.
[6] DOUGLAS A, CARTER R, OAKES L, SHARE K, COHN A P, PINT C L. Ultrafine iron pyrite (FeS2) nanocrystals improve sodium–sulfur and lithium–sulfur conversion reactions for efficient batteries[J]. ACS Nano, 2015, 9(11):11156−11165.
[7] WALTER M, KRAVCHYK K V, IBANEZ M, KOVALENKO M V. Efficient and inexpensive sodium–magnesium hybrid battery[J]. Chemistry Material, 2015, 27(21): 7452−7458.
[8] CAO F, PAN G X, CHEN J, ZHANG Y J, XIA X H. Synthesis of pyrite/carbon shells on cobalt nanowires forming core/branch arrays as high-performance cathode for lithium ion batteries[J]. Journal of Power Sources, 2016, 303(30): 35−40.
[9] QIU Wen-da, XIA Jian, ZHONG Hai-min, HE Shen-xian, LAI Shu-hui, CHEN Liu-ping. L-cysteine-assisted synthesis of cubic pyrite/nitrogen-doped graphene composite as anode material for lithium-ion batteries[J]. Electrochimica Acta, 2014, 137(10): 197−205.
[10] 黎佩珊, 孙明, 郑育英, 徐前忠. 不同纯度的正极材料黄铁矿的表征及其性能研究[J]. 广东化工, 2012, 39(16): 1−2. LI Pei-shan, SUN Ming, ZHENG Yu-ying, XU Qian-zhong. Characterization and electritic properties of pyrite with different purity[J]. Guangdong Chemical, 2012, 39(16): 1−2.
[11] 王大刚, 范力仁, 王圣平, 范畴, 何明中, 栗海峰. 黄铁矿作为锂电池正极材料的电化学性能[J]. 材料导报, 2012, 26(9): 93−96. WANG Da-gang, FAN Li-ren, WANG Sheng-ping, FAN Chou, HE Ming-zhong, LI Hai-feng. Electrochemical properties of pyrite as lithium battery cathode materials[J]. Materials Review, 2012, 26(9): 93−96.
[12] 杨兆堂, 刘效疆. 水热法合成FeS2材料及其在热电池中的性能[J]. 材料保护, 2013, 46(S1): 51−53. YANG Zhao-tang, LIU Xiao-jiang. Preparing pyrite via hydrothermal method for thermal battery applications[J]. Materials Protection, 2013, 46(S1): 51−53.
[13] 龙 飞, 神 征, 莫淑一, 邹正光. 黄铁矿(FeS2)薄膜制备和掺杂改性的研究进展[J]. 半导体光电, 2015, 36(6): 857−862. LONG Fei, SHEN Zheng, MO Shu-yi, ZOU Zheng-guang. Research progress on deposition and doping techniques of FeS2thin film[J]. Semiconductor Optoelectronics, 2015, 36(6): 857−862.
[14] CHATZITHEODOROU G, FIECHTER S, KONENKAMP R, KUNST M, JAEGERMANN W, TRIBUTSCH H. Thin photoactive FeS2(pyrite) films[J]. Materials Research Bulletin, 1986, 21(12): 1481−1487.
[15] LEHNER S W, NEWMAN N, van SCHILFGAARDE M, BANDYOPADHYAY S, SAVAGE K, BUSECK P R. Defect energy levels and electronic behavior of Ni-, Co-, and As-doped synthetic pyrite (FeS2)[J]. Journal of Applied Physics, 2012, 111(8): 083717-1−083717-7.
[16] FERRER I J, de las HERAS C, SANCHEZ C. Physical properties of Cu-doped FeS2pyrite thin films[J]. Applied Surface Science, 1993, 70(2): 588−592.
[17] YU Qi-kai, CAI Shu, JIN Zheng-guo, YAN Zi-peng . Evolutions of composition, microstructure and optical properties of Mn-doped pyrite (FeS2) films prepared by chemical bath deposition[J]. Materials Research Bulletin, 2013, 48(9): 3601−3606.
[18] 李玉琼, 陈建华, 陈 晔, 郭 进. 黄铁矿(100)表面性质的密度泛函理论计算及其对浮选的影响[J]. 中国有色金属学报, 2011, 21(4): 919−926. LI Yu-qiong, CHEN Jian-hua, CHEN Ye, GUO Jin. Density functional theory calculation of surface properties of pyrite (100) with implications for flotation[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(4): 919−926.
[19] CHEN Jian-hua, CHEN Ye, LI Yu-qiong. Effect of vacancy defects on electronic properties and activation of sphalerite(110) surface by first-principles[J]. Transactions of Nonferrous Metals Society of China, 2010, 21(3): 502−506.
[20] 陈建华, 李玉琼, 衷水平, 郭 进. 含空位缺陷黄铁矿(100)表面吸附氢氧根和羟基钙的量子化学研究[J]. 中国有色金属学报, 2013, 23(3): 859−865. CHEN Jian-hua, LI Yu-qiong, ZHONG Shui-ping, GUO Jin. Quantum chemical study of adsorption of hydroxyl and hydroxyl calcium on pyrite (100) surface bearing vacancy defects[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(3): 859−865.
[21] 陈建华, 钟建莲, 李玉琼, 陈 晔, 郭 进. 黄铁矿、白铁矿和磁黄铁矿的电子结构及可浮性[J]. 中国有色金属学报, 2011, 21(7): 1719−1727. CHEN Jian-hua, ZHONG Jian-lian, LI Yu-qiong, CHEN Ye, GUO Jin. Electronic structures and floatability of pyrite, marcasite and pyrrhotite[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(7): 1719−1727.
[22] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J, PROBERT M J, REFSON K, PAYNE M C. First principles methods using CASTEP[J]. Zeitischrift fuer Kristallographie, 2005, 220(5/6): 567−570.
[23] VANDERBILT D. Soft self-consistent pseudopotentials in generalized eigenvalue formalism[J]. Physical Review B, 1990, 41(11): 7892−7895.
[24] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188−5192.
[25] PRINCE K C, MATTEUCCI M, KUEPPER K, CHIUZBAIAN S G, BARKOWSKI S, NEUMANN M. Core-level spectroscopic study of FeO and FeS2[J]. Physical Review B, 2005, 71(8): 085102-1−085102-9.
[26] OERTZEN G U , JONES R T, GERSON A R. Electronic and optical properties of Fe, Zn and Pb sulfides[J]. Physics and Chemistry of Minerals, 2005, 32(4): 255−268.
[27] 陈建华. 硫化矿物浮选固体物理研究[M]. 长沙: 中南大学出版社, 2015. CHEN Jian-hua. The solide physics of sulphide minerals flotation[M]. Changsha: Central South University Press, 2015.
[28] JONES R O, GUNNARSSON O. The density functional formalism, its applications and prospects[J]. Review of Modern Physics, 1989, 61(3): 689−746.
Effect of As doping concentration on electronic structure and optical properties of FeS2
MA Wan-kun1, CHEN Jian-hua2, ZHANG Guo-fan1, LI Yu-qiong2, FENG Qi-ming1
(1. School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China;2. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China)
Aimed at verifying the effect of As doping concentration on FeS2, geometrical structure, electronic structure and optical properties of As-doped FeS2were calculated using pseudo-potential plane-wave method of the first principle based on the density function theory. Three supercells (2×2×2, 2×2×1 and 2×1×1) were introduced to achieve different doping concentrations (1.93%, 3.82% and 7.48%, mass fraction). The results of geometrical structure, electronic structure show that lattice constant and volume of unit cell increase with As addition, the conduction band moves negatively while the width of forbidden band decreases. Moreover, the density of states in the range of −10.4 − −9.5 eV exhibits impurity level composed of As p. The calculation of optical properties reveals the static dielectric constant, refractive index and photo conductivity aggrandize with the ever-increasing doping concentration, which indicates FeS2doped As possesses stronger absorption capacity of light and higher photoelectric conversion efficiency. Therefore, the calculation lays some theory foundation for exploitation and development of FeS2photoelectric materials.
As-doping; pyrite; optical and electrical property; first principle
(编辑 何学锋)
Project(2014CB643402) supported by National Basic Research Development Program of China; Project (51304054) supported by the National Natural Science Foundation of China; Project (2013GXNSFBA019259) supported by the Guangxi Natural Science Foundation, China
2016-02-24; Accepted date:2016-07-05
ZHANG Guo-fan; Tel: +86-731-88830913; E-mail: zhangguofan2002@qq.com
1004-0609(2017)-02-0414-09
O472
A
国家重点基础研究发展规划资助项目(2014CB643402);国家自然科学基金资助项目(51304054);广西自然科学基金资助项目(2013GXNSFBA019259)
2016-02-24;
2016-07-05
张国范,教授,博士;电话:0731-88830913;E-mail: zhangguofan2002@qq.com
