基于QbD理念的安宫牛黄丸整体混合终点评价方法研究
2017-04-05刘晓娜郑秋生车晓青吴志生乔延江
刘晓娜+郑秋生+车晓青+吴志生+乔延江
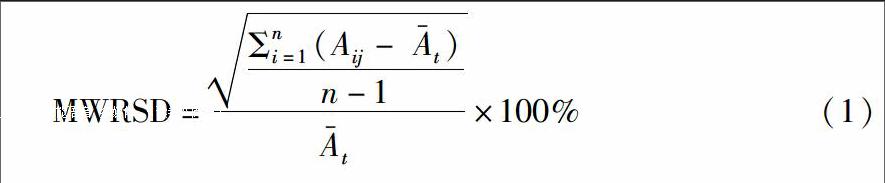
[摘要]大品种中药安宫牛黄丸混合终点判断是关键的技术问题。基于质量源于设计(QbD)理念的控制策略,研究提出一种整体混合终点的判断方法,为含矿物质中药混合提供方法学。采用激光诱导击穿光谱(LIBS)技术获得安宫牛黄丸混合中间体的光谱数据。结合移动窗相对标准偏差法(MWRSD)法,朱砂、雄黄和珍珠粉为整合研究指标,通过比较相邻混合时间光谱的差异,快速评价整体混合过程。雄黄、朱砂和珍珠粉3个药味的混合过程并非完全一致,但在混合的最后一个阶段,3个药味均达到了混合均匀;得到建议混合终点。采用LIBS过程技术建立的微区时序分析方法,实施过程控制。所建立的方法无需标准光谱库,具有分析快速、近似无损、无需复杂的样品前处理的等特点,为大品种中药制剂的质量研究提供新思路。
[关键词]安宫牛黄丸; 混合终点; 激光诱导击穿光谱; 质量源于设计; 过程分析技术
[Abstract]The blending endpoint determination of Angong Niuhuang Wan (AGNH) is a key technology problem The control strategy based on quality by design (QbD) concept proposes a whole blending endpoint determination method, and provides a methodology for blending the Chinese materia medica containing mineral substances Based on QbD concept, the laser induced breakdown spectroscopy (LIBS) was used to assess the cinnabar, realgar and pearl powder blending of AGNH in a pilotscale experiment, especially the whole blending endpoint in this study The blending variability of three mineral medicines including cinnabar, realgar and pearl powder, was measured by moving window relative standard deviation (MWRSD) based on LIBS The time profiles of realgar and pearl powder did not produce consistent results completely, but all of them reached even blending at the last blending stage, so that the whole proposal blending end point was determined LIBS is a promising Process Analytical Technology (PAT) for process control Unlike other elemental determination technologies such ICPOES, LIBS does not need an elaborate digestion procedure, which is a promising and rapid technique to understand the blending process of Chinese materia medica (CMM) containing cinnabar, realgar and other mineral traditional Chinese medicine This study proposed a novel method for the research of large varieties of traditional Chinese medicines
[Key words]Angong Niuhuang Wan; whole blending endpoint; laserinduced breakdown spectroscopy; quality by design; process analytical technology
安宮牛黄丸是中医治疗高热症的“温病三宝”之一[1],历来是中医用于治疗各类急证的必备要药。安宫牛黄丸处方的组分比较复杂,其中雄黄、朱砂比例均为10%,药味的比重差异较大,从而影响药效和安全性。粉末混合过程为动态变化的时间序列,其主要目的是确定混合终点,指导混合工艺优化[23]。大品种中药安宫牛黄丸混合终点判断是关键的技术问题。马群[4]应用近红外在线分析技术检测安宫牛黄丸原粉混合终点,运用相似度匹配法判断混合终点;刘珊珊等[5]采用近红外光谱结合移动窗标准偏差法对安宫牛黄丸的中试混合过程进行在线监测;并采用高效液相色谱法验证中间体中有效成分黄芩苷等成分的变化,验证了近红外在线分析技术指导混合终点判断的可靠性。目前尚未有针对矿物质中药的混合过程的研究,缺乏质量控制的有效方法,难以保证含矿物质中成药质量的稳定、均一。21世纪初,美国推出了Quality by Design“QbD(质量源于设计)”理念。QbD 是一种系统的研究方法,强调对产品和工艺的理解,以及对工艺过程的控制[67]。在制药行业,QbD的初始设计决定产品质量的理念已逐渐被业界认可。“质量可控、安全有效”是药品研发过程须遵循的首要原则。其中,质量可控又是安全有效的前提条件。激光诱导击穿光谱技术(laserinduced breakdown spectroscopy,LIBS)是新兴的微区多元素检测技术,是以激光脉冲作为激发源诱导产生激光等离子体的原子发射光谱[810]。在解决多指标、多成分的中药分析时,LIBS区别于传统的元素分析技术,具有诸多的优势:快速、绿色、多元素检测及近似无损的特点[11]。本研究基于QbD理念,采用LIBS过程分析技术评价安宫牛黄丸的整体混合过程,以朱砂、雄黄和珍珠粉3个药味为研究载体,通过LIBS技术在若干时间点代表性的采样获得混合过程的微区光谱。以特定元素砷(As)、汞(Hg)、钙(Ca)分析结果为断面,快速评价安宫牛黄丸的混合过程的微区时序特征,了解含矿物质中药粉末混合过程的规律,建立合理的质量评价方法,指导整合终点判断;确保含矿物质中药混合的均一性,为保证生产过程的工艺可控性提供了基础。
1材料
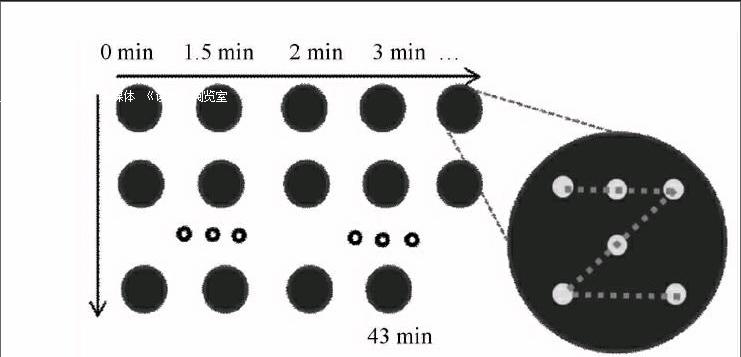
黄芩、黄连、栀子、郁金、牛黄、水牛角浓缩粉、朱砂、雄黄、冰片和珍珠粉等药材均由北京同仁堂股份有限公司科学研究所提供。安宫牛黄丸混合中间体由北京中医药大学马群教授提供。模拟安宫牛黄丸的混合过程,并得到44个安宫牛黄丸混合中间体样品(0,15,2,3,4,5 min,每隔1 min取样至43 min)。
激光诱导击穿光谱采用LIBS实验系统,由北京市农林科学院,国家农业信息化工程技术研究中心友情提供;系统由激光器、光谱仪、三维精密运动平台、CCD探测器、信号延时器组成。光谱仪采用海洋光学公司(Ocean Optics)的HR 2000+,波长范围198~876 nm。FW4/A红外压片机,天津市新天光分析仪器技术有限公司。
2方法
21安宫牛黄丸混合中间体的LIBS 光谱采集
为防止激光击打时的粉末飞溅现象,分别取安宫牛黄丸混合中间体适量,采用红外压片机压制呈直径13 mm、厚度约1 mm的锭片,共制备44个锭片样品。光谱采集方式见图1,计算6个采样位点的平均光谱,即得到混合过程的微区时序光谱。
22LIBS差谱法
珍珠的主要成分有碳酸钙。由于钙信号同时来自其他药味,故采用差谱法归属珍珠粉中Ca特征谱线。
23安宫牛黄丸混合终点判断方法
采用移动窗标准相对偏差(moving window relative standard deviation,MWRSD)[1213]进行中间体整合终点判断。MWRSD选择混合中间体的n张连续的光谱,计算n张连续光谱中待测元素的特征谱线强度的相对标准偏差(RSD),剔除原n 条光谱中时间最早的光谱,补充一条新光谱,重新计算标准偏差,以此类推计算所有时序光谱数据,计算公式如下。
Ai,Aij分别为i,ij时混合中间体的LIBS光谱中砷元素或汞元素的相对信号强度。此处选择移动窗口n=3;每次增加1个样品,连续移动。最终得到混合过程中不同取样点中间体样品光谱的标准偏差。以RSD为纵坐标,混合时间为横坐标作图。RSD越小,表明元素的特征谱线信号强度变化小,混合越均匀,提示混合终点。
刘晓娜等:基于QbD理念的安宫牛黄丸整体混合终点评价方法研究24数据处理和软件
采用OOILIBS (Ocean Optics) 软件读取光谱数据;采用ORIGIN 8软件画图。
3结果
31安宫牛黄丸混合中间体的LIBS光谱
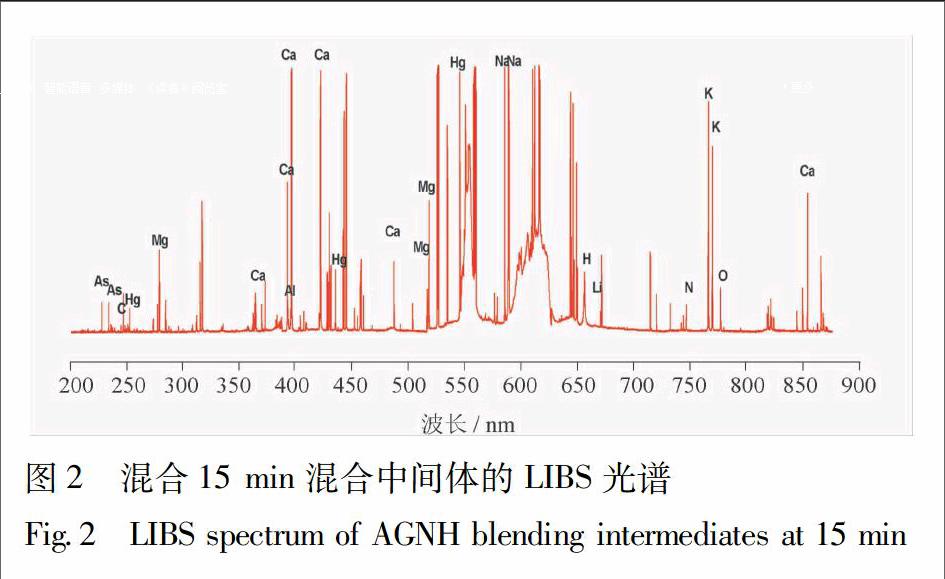
混合过程中15 min混合中间体的LIBS 光谱见图2。混合中間体的特征谱线除了Ca,钾(K),钠(Na),镁(Mg),锂(Li),碳(C),氢(H),氧(O),氮(N),等;同时出现As(228816,234984,616979 nm)和Hg(253651,296732,313123 nm)。谱中590~610 nm处受到基质效应影响,但并未影响As,Hg的特征谱线。225~260 nm的局部放大图见图3,As 228816 nm,Hg 253651 nm处特征谱线未受到其他元素的干扰[1415]。因此,选择此处特征谱线,研究安宫牛黄丸混合过程中矿物药雄黄、朱砂的变化规律。
32Ca的LIBS差谱
Ca的特征谱线有315839,370627,373668,393379,396829,422640,430228,487704,534970,558842,585802,612715,616231,643965,646284,649403,714852,720267,732426,849760,849760,854172 nm [1618]。Ca同时来自于植物药材和珍珠粉,故将30 min 混合中间体LIBS 谱与0 min 谱做差谱,然后比对0 min 样本与差谱之间的光谱差异,见图4,最终选定特征谱线393379,585746,643965,854172 nm为珍珠粉中Ca的光谱信号。
33安宫牛黄丸整体混合终点判断
331朱砂和雄黄混合终点判断采用MWRSD法对LIBS实验数据进行处理,设定10%为阈值。将混合过程进行划分时段,As和Hg在不同的特征谱线处的MWRSD变化趋势图见图5。图5 (a) 划分4个阶段,在混合的前20 min,Hg 53651 nm处的MWRSD变化剧烈;随着混合的进行,B段(20~27 min)MWRSD相对较平稳,且MWRSD小于10%;进一步混合,C段(27~37 min)中MWRSD出现较大幅度波动;D段(37~43 min)MWRSD呈相对平稳阶段且MWRSD小于10%,提示到达混合终点到达。与Hg相似,As 228816 nm的混合可以划分了4个阶段,见图5 (b)。A段(0~27 min)和C段(32~38 min)过程中MWRSD变化剧烈;B段(21~28 min)和D段(38~43 min)过程中MWRSD保持相对平稳,且MWRSD小于10%。虽然As和Hg在B段均出现相对平稳显现,但混合并不完全,并非混合终点。综合考虑,As和Hg的混合终点D段,建议混合终点为38 min。
332珍珠粉的混合终点判断珍珠粉是安宫牛黄丸的组成成分之一,其主要成分是碳酸钙。珍珠粉的混合是否与朱砂、雄黄的混合过程一致,混合终点是否一致?研究探讨安宫牛黄丸混合过程中,珍珠粉的混合过程变化特点和规律。混合过程中Ca在不同的特征谱线处的MWRSD变化趋势图见图6。图6 (a) 划分了4个阶段,在混合的前20 min,Ca 393379 nm处的MWRSD变化剧烈;随着混合的进行,B段(20~29 min)MWRSD相对平稳,MWRSD小于10%;进一步混合,C段(29~38 min)中MWRSD出现波动;D段(38~43 min)MWRSD呈相对平稳阶段且MWRSD小于10%,提示到达混合终点。图6 (b) 显示2个混合阶段,A段 在混合开始的前13 min,Ca 585746 nm处的MWRSD变化剧烈,提示激烈混合的过程;B段在混合13~43 min过程中MWRSD相对平稳,且均小于10%,表明在此阶段混合较平稳。图6(c)中Ca 643965 nm处的MWRSD变化趋势图与图6 (a) Ca 393379 nm处的MWRSD变化趋势图相似,混合分为4个阶段;A段(0~21 min)和C段(27~38 min)过程中MWRSD变化剧烈;B段(21~28 min)和D段(38~43 min)过程中MWRSD保持相对平稳,而且MWRSD小于10%。与图6 (a)、图6(c)相似,图6(d)中Ca 854172 nm处的MWRSD变化同样呈现4个阶段,在混合开始的13 min,MWRSD变化剧烈,随着混合的进行,B段(13~27 min)内MWRSD变化平稳,进一步混合,在混合的27~38 min 时MWRSD出现微小波动,在38 min 后(D段)MWRSD相对平稳且小于10%。Ca在4个特征谱线处处特征谱线的建议混合终点为38 min。
3333个药味整体的混合终点判断LIBS技术结合MWRSD的朱砂、雄黄和珍珠粉在B段和D段的微区时序混合分析结果见图 7。对比发现在B段 Hg与Ca共同时间段为21~27 min;但是As即雄黄在B段混合相对均匀时间段为27~31 min;表明在B段雄黄与朱砂和珍珠粉的混合并非同步。在D段(Ca 585746 nm为D段),其他元素共同时间段为38~43 min,表明3个药味同时达到了混合均匀;故朱砂、雄黄和珍珠粉的共同混合终点为38 min。
4讨论
QbD理念正推动着药品生产模式从传统的以检验为主到以科学地研究生产过程为主。ICH发布
的Q8(pharmaceutical development,Q8)药物研发中指出,质量是通过设计赋予的。获得合格的质量,必须加强对产品的理解和对成产的全过程控制。ICH Q8的控制策略为“源于对现行产品和工艺的理解,制定一系列有计划的控制,用于确保工艺性能与产品质量的[6]。所有产品的质量属性和工艺参数,均属于ICH Q8控制策略范畴。QbD理念,要求生产过程中对工艺过程进行“实时质量保证”,保证每个步骤工艺的输出均符合质量要求。关键工艺参数合格即可保证产品质量达到要求。
粉末混合过程是中药固体制剂生产的重要操作单元之一,也是时序过程。药物混合均匀度直接影响药品的疗效。药物混合均匀度直接影响药品的疗效。安宫牛黄丸是一种完全以药材原粉入药的中药丸剂,其处方的组分比较复杂,植物性药材、动物性药材和矿物性药材同时出现在处方中,导致药味的比重差异较大,尤其是含重金属矿物质如朱砂、雄黄等中药制剂的混合均匀与否不仅影响其疗效,而且严重影响其用药安全。对于中药的混合过程的评价多采用离线液相色谱法、气相色谱法等对其判断。这些方法虽然准确度高,但样品处理复杂,且多以研究有机化合物为主,未考虑矿物质中药的混合特征。
本文基于QbD控制策略,将LIBS过程技术应用于安宫牛黄丸的整体混合过程,结合MWRSD法研究雄黄、朱砂和珍珠粉3个药味的微区时序特征。构建的金属元素快速检测方法,实现整体混合终点的快速判断,确保混合均匀。结果表明:在混合的整个时序过程中,初始阶段物料混合相对剧烈,混合效率高,到達最佳的混合状态后反方向变换,出现偏析或分料现象;随着进一步混合,颗粒以扩散混合为主。在整个混合过程中物料的混合和分离同时发生,当二者达到动态平衡的状态即混合终点。研究发现物料混合均匀的时间不是一个固定的时间点,而且是一个时间段,超过此时间段,粉末会出现过混合现象。雄黄、朱砂和珍珠粉的混合过程并非完全一致,但在混合的最后一个阶段,3个药味均达到了混合均匀;得到建议混合终点,且无过混合现象发生。所建立的方法无需建立标准光谱库,通过比较时间序列中相邻混合时间的光谱差异,实现了朱砂、雄黄和珍珠粉混合终点的快速判断。
过程分析技术(PAT)属于第三级控制策略,其目的在于增加过程理解、提高生产效率和产品质量,其优势在于实时分析、监测、控制。LIBS技术属于PAT技术,在微区时序分析研究方面彰显了其快速、实时、近似无损等优势,有望用于中药混合过程的回馈控制,有效避免过度混合和资源浪费。将PAT技术应用于中药材粉末的混合过程,研究混合过程时序变化的特点和规律,对过程理解具有重要的意义,同时为含矿物质中药制剂混合的在线监测提供了技术支持。
[参考文献]
[1]王欣美, 张甦, 王枚博, 等 安宫牛黄丸体外安全性评价方法的研究[J] 光谱学与光谱分析, 2015, 35(1): 238
[2]薛忠, 徐冰, 张志强, 等 药物粉末混合过程在线监控技术研究进展[J] 中国药学杂志, 2016, 51(2): 91
[3]杨婵, 徐冰, 张志强, 等 基于移动窗F检验法的中药配方颗粒混合均匀度近红外分析研究[J] 中国中药杂志, 2016, 41(19): 3557
[4]马群 安宫牛黄丸生产过程质量评价方法研究[D] 北京: 北京中医药大学, 2007
[5]刘倩. 中药粉末混合过程分析和中试放大效应研究[D]. 北京:北京中医药大学, 2014.
[6]徐冰, 史新元, 乔延江, 等 中药制剂生产工艺设计空间的建立[J]中国中药杂志, 2013, 38(6): 1001
[7]刘倩 中药粉末混合过程分析和中试放大效应研究[D] 北京:北京中医药大学, 2014
[8]Hahn D W, Omenetto N Laserinduced breakdown spectroscopy (LIBS), part I: review of basic diagnostics and plasmaparticle interactions: stillchallenging issues within the analytical plasma community[J] Appl Spectrosc, 2010, 64(12): 335
[9]Hahn D W, Omenetto N Laserinduced breakdown spectroscopy (LIBS), part Ⅱ: review of instrumental and methodogical approaches to material analysis and applications to different fields[J] Appl Spectrosc, 2012, 66(4): 347
[10]Singh V K, Rai A K Prospects for laserinduced breakdown spectroscopy for biomedical applications: a review[J] Laser Med Sci, 2011, 26 (5): 673
[11]Cremers D A, Chinni R C Laserinduced breakdown spectroscopy—capabilities and limitations[J] Appl Spectrosc Rev, 2009, 44(6): 457
[12]Blanco M, GozálezBaó R, Bertran E Monitoring powder blending in pharmaceutical processes by use of near infrared spectroscopy[J] Talanta, 2002, 56: 203
[13]Scheibelhofer O, Balak N, Wahl P R, et al Monitoring blending of pharmaceutical powders with multipoint NIR spectroscopy[J] AAPS Pharm Sci Tech, 2013, 14(1): 234
[14]Kwak J H, Lenth C, Salb C, et al Quantitative analysis of arsenic in mine tailing soils using double pulselaser induced breakdown spectroscopy[J] Spectrochimica Acta Part B, 2009, 64: 1105
[15]Fang X, Ahmad S R Detection of mercury in water by laserinduced breakdown spectroscopy with sample preconcentration[J]. Appl Phys B, 2012, 106: 453
[16]Anzano J, Lasheras R J Strategies for the identification of urinary calculus by laser induced breakdown spectroscopy [J] Talanta, 2009, 79(2): 352
[17]Kurniawan K H, Tjia M O, Kagawa K Review of laserinduced plasma, its mechanism, and application to quantitative analysis of hydrogen and deuterium [J]Appl Spectrosc Rev, 2014, 49(5): 323
[18]Bahreini M, Ashrafkhani B, Tavassoli S H Elemental analysis of fingernail of alcoholic and doping subjects by laserinduced breakdown spectroscopy[J] Appl Phys B Lasers Opt, 2014, 114(3): 439
[19]Liu Xiaona, Ma Qun, Liu Shanshan, et al Monitoring As and Hg variation in AnGongNiuHuang Wan (AGNH) intermediates in a pilot scale blending process using laserinduced breakdown spectroscopy[J] Spectrochim Acta Pt A Mol Bio, 2015, 15: 1547[責任编辑孔晶晶]
