铁铁氢化酶模型配合物的合成及其电催化产氢行为研究
2017-03-30高尚张唯一杨婷婷
高尚,张唯一,杨婷婷
(长春理工大学材料科学与工程学院,长春 130022)
铁铁氢化酶模型配合物的合成及其电催化产氢行为研究
高尚,张唯一,杨婷婷
(长春理工大学材料科学与工程学院,长春 130022)
通过十二羰基三铁与2,3-二巯基吡嗪在四氢呋喃中反应将刚性共轭桥引入到铁铁氢化酶活性中心模型配合物中,化学模拟合成了[μ-SC4N2H2S-μ]Fe2(CO)6(配合物I)。研究了配合物I的PMe3配体取代过程,得到双核铁配合物[μ-SC4N2H2S-μ]Fe2(CO)5(PMe3)(II)和单核铁配合物[μ-SC4N2H2S-μ]Fe(CO)2(PMe3)2(III)。电化学循环伏安研究表明吸电子共轭桥连结构能有效降低[2Fe2S]模型配合物中心铁原子上的电子云密度及其还原电位。配合物I相应的第一还原电位出现在-1.18V(vs.Fc/Fc+),是目前已见报道中最正的。同时,配合物I、II、III在不同的还原电位下均具有电催化质子还原产氢的活性。其中,配合物III在-2.28V(vs.Fc/Fc+)下的电催化活性最高。
氢化酶;化学模拟;循环伏安法;电催化
氢气是一种清洁、高效的能源载体[1],随着人们对能源的需求急剧增加,开发低成本的产氢催化剂成为备受瞩目的研究课题。自然界中,厌氧微生物体内存在一种能可逆催化质子还原产氢的酶,被称为氢化酶[2]。根据其活性中心金属的不同,可分为三类:[FeFe]-氢化酶、[NiFe]-氢化酶和[Fe]-氢化酶[3]。其中,[FeFe]-氢化酶具有较高的催化活性,每摩尔活性中心每秒可产生6000~9000分子氢气[4],从而备受关注。[FeFe]-氢化酶活性中心由一个[4Fe4S]立方烷通过半胱氨酸硫原子与一个[2Fe2S]单元相连(图1)。在催化质子还原过程中,[4Fe4S]簇通常作为传递电子的通道,而[2Fe2S]单元是催化反应的活性部位,负责吸附质子,生成氢气。[2Fe2S]单元呈蝶状结构,与铁原子配位的除[4Fe4S]簇外还有CN-和CO两类配体[5,6]。氢化酶活性中心结构的揭示,为人们在分子水平上研制成本低廉的高效产氢催化剂奠定了基础。氢化酶活性中心的化学模拟也成为生物无机化学[7]领域的研究热点。
近年来,科研工作者合成了大量的[2Fe2S]及[2Fe3S]氢化酶活性中心结构和功能模型,广泛地探索了其催化质子还原产氢性能,在模型配合物电催化产氢方面取得了突破性进展[8-10]。但是,目前合成的全羰基[2Fe2S]配合物在乙腈溶液中的第一还原电位普遍较高(-1.5~-1.8V vs.Fc/Fc+),当分子中一个或两个羰基被供电性配体取代后,其还原电位向负方向移动至-1.9~-2.7V(vs.Fc/Fc+),与自然界中(-0.4V vs.NHE)相去甚远。最近,有研究发现:当两个硫原子之间以苯环连接时,模型配合物具有较低的第一还原电位(-1.44V vs.Fc/Fc+)[11]。可见,刚性共轭桥连结构有利于降低[2Fe2S]配合物的还原电位。

图1 [FeFe]-氢化酶活性中心化学结构图(中)及模型配合物I、II(左)、III(右)分子结构图
受此启发,将吡嗪环作为桥连配体,合成了配合物[(μ-SC4N2H2S-μ)Fe2(CO)6](I,图1),成功地将[2Fe2S]配合物的第一还原电位降低至-1.18V vs.Fc/Fc+。在对配合物I进行PMe3配体取代研究的过程中,得到了双核铁单PMe3配体取代配合物[(μ-SC4N2H2S-μ)Fe2(CO)5(PMe3)](II,图1),及单核铁双PMe3取代配合物[(μ-SC4N2H2S-μ)Fe(CO)2(PMe3)2](III,图1)[12]。下面将着重阐述配合物I、II和III的合成及电化学行为研究。
1 实验
1.1 仪器与试剂
仪器:Varian INOVA 400核磁共振仪;Q-Tof Micro高分辨质谱仪;Thermo Nicolet Nexus中/远红外气相色谱-傅立叶变换红外光谱联用仪;BAS-100B电化学工作站。
试剂:十二羰基三铁;2,3-二氯吡嗪;NaHS;三甲基氧化胺;三甲基膦。所有溶剂使用前均按照标准方法进行除水除氧处理。
1.2 合成与表征
配合物I的合成:将Fe3(CO)12(5.75g,11.4mmol)和2,3-二巯基吡嗪(2.16g,15mmol)溶解到100mL四氢呋喃中,氮气保护下回流搅拌2小时。减压蒸干溶剂,在硅胶柱上用正己烷/二氯甲烷作为洗脱剂分离提纯。得到红色固体(2.57g),收率:40%。1H NMR(400MHz,CDCl3):δ=7.58(s,2H,C4N2H2)ppm;13C NMR(100MHz,CDCl3):δ=206.3,169.2,137.3ppm;IR(CH2Cl2):nmax/cm-12085,2045,2017,1998(CO);HR-MS(EI):m/z calc.for[M+]:421.8053;found:421.8063.
配合物II的合成:N2保护下,向配合物I(0.42 g,1.0mmol)的乙腈溶液中一次加入三甲基氧化胺(0.11g,1.1mmol),反应液室温搅拌10分钟后变为深红色后加入三甲基膦(0.08g,1.0mmol),搅拌20分钟。在中性氧化铝柱上用正己烷/二氯甲烷作为洗脱剂分离提纯。收集第一个红色谱带,蒸干溶剂,得到红色固体(0.04g),收率:10%。1H NMR(400MHz,CDCl3):δ=7.46(s,2H,C4N2H2),1.60(s,9H,PMe3)ppm;31P NMR(CDCl3):δ=20.7ppm;IR(CH2Cl2):nmax/cm-12050,1986,1936(CO);HRMS(EI):m/z calc.for[M+]:469.8546;found:469.8578.
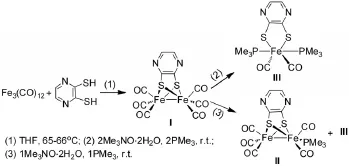
图2 模型配合物I、II、III的合成路线
配合物III的合成:N2保护下,向配合物I(0.42g,1.0mmol)的乙腈溶液中一次加入三甲基氧化胺(0.24g,2.2mmol),反应液室温搅拌10分钟后变为深红色后加入三甲基膦(0.15g,2.0mmol),搅拌20分钟。在中性氧化铝柱上用正己烷/二氯甲烷作为洗脱剂分离提纯。收集主要的橙红色谱带,蒸干溶剂,得到橙红色固体(0.18g),收率:45%。1H NMR(400MHz,CDCl3):δ=7.68(s,2H,C4N2H2),1.50(s,18H,PMe3)ppm;13C NMR(100MHz,CDCl3):δ=210.6,167.2,135.1,14.7(t)ppm;31P NMR(CDCl3):δ=8.5ppm;IR(CH2Cl2):nmax/cm-12016,1959(CO);HR-MS(ESI):m/z calc.for[M+H+]:406.9869;found:406.9872.
2 结果与讨论
2.1 合成
利用2,3-二巯基吡嗪与十二羰基三铁在四氢呋喃溶液中回流即可得到配合物I,经硅胶柱分离得到的产物为深红色固体,可溶于多种有机溶剂,且在空气中稳定存在。在对I进行PMe3配体取代研究的过程中,首先利用I与二倍当量的PMe3配体在甲苯溶液中回流,原料反应完全后,硅胶/氧化铝柱分离得到橙色固体,经红外、核磁、高分辨质谱表征证实为单核铁六配位结构的配合物III。为了消除温度的影响,又采用Me3NO·2H2O作为脱羰基试剂,PMe3配体室温取代,依然只得到III。而在对I进行单PMe3配体取代的过程中,除了生成少量单PMe3取代[2Fe2S]配合物II,主要产物仍为III。可见,配合物III是远比双PMe3取代配合物[(μ-SC4N2H2S-μ)Fe2(CO)4(PMe3)2]和双核铁单PMe3取代配合物II稳定的物种。

表1 配合物I、II、III及相关[2Fe2S]配合物的红外吸收峰
表1为配合物I、II、III与其他相关[2Fe2S]配合物的羰基红外吸收数据,羰基红外吸收峰的位移可以反映配合物中心铁原子上电子云密度的高低。与苯环桥[2Fe2S]配合物[(μ-bdt)Fe2(CO)6]相比,配合物I羰基吸收峰的平均值红移了7cm-1;单核铁双PMe3取代配合物III,其羰基吸收峰的平均值相对于I向低波数移动了49cm-1,远小于[(μ-pdt)Fe2(CO)6]发生双PMe3取代时的红移值。单PMe3取代配合物II羰基吸收峰的平均值相对于I红移了45cm-1,也要小于[(μ-pdt)Fe2(CO)6]发生单PMe3取代时的红移值。
2.2 循环伏安曲线
以n-Bu4NPF6为支持电解质,在乙腈溶液中测量了配合物I、II、III的电化学行为(图3),测试过程中电位扫描方向为负方向,扫描速率为100mV s-1,所有电位均相对于二茂铁的半波电位而言。
配合物I在+0.89V(vs.Fc/Fc+)呈现出一个不可逆的氧化峰,可以归属为FeIFeI→FeIFeII的单电子氧化过程;在-1.18V(vs.Fc/Fc+)出现第一还原峰,对应着FeIFeI→FeIFe0的还原过程。配合物I的第一还原电位比丙烷桥及苯环桥联的[2Fe2S]配合物[(μ-pdt)Fe2(CO)6]和[(μ-bdt)Fe2(CO)6]相应的还原电位分别向阳极方向移动470及250mV,为目前在全羰基[2Fe2S]配合物中发现的最正的还原电位,说明吡嗪环上氮杂原子的吸电子性可有效降低铁原子上的电子云密度。另外,配合物I的还原峰有着更为优越的可逆性,根据文献推测,配合物I的还原生成了一种含桥羰基的稳定结构[16]。
配合物I中的一个羰基由PMe3取代后所得的配合物II,其第一还原电位相对于I向阴极方向移动,这是因为PMe3的配位增大了铁原子上的电子云密度所致。配合物III在-2.02V和-2.28V(vs.Fc/ Fc+)出现两个还原峰,双PMe3配体的引入进一步增大了中心铁原子上的电子云密度,因此配合物III的还原峰出现在更负的位置。配合物III中的单核铁为+2价,测量所得的两个还原峰分别对应着FeII→FeI和FeI→Fe0的还原过程。

图3 模型配合物I、II、III的循环伏安曲线
2.3 电催化性能
配合物I、II、III在乙酸(HOAc)存在下的循环伏安曲线如图4所示。乙酸的加入并未使配合物I在-1.18V处的还原峰电流出现明显变化,说明配合物I在第一还原电位处不具备电催化质子还原的活性。而随着HOAc浓度的增加,配合物I在-2.21V处的还原峰电流呈线性增长,且向阴极方向有规律的移动;这是典型的电化学催化质子还原产氢行为的特征现象。另外,在该电位下电解时,气相色谱监测到有氢气生成,进一步证实配合物I在-2.21V处的第二还原峰具有电催化质子还原的活性。相同条件下,配合物II的电催化质子还原产氢电位为-1.72V;而单核铁配合物III在-2.02、-2.28V处均具备电催化质子还原产氢的活性。
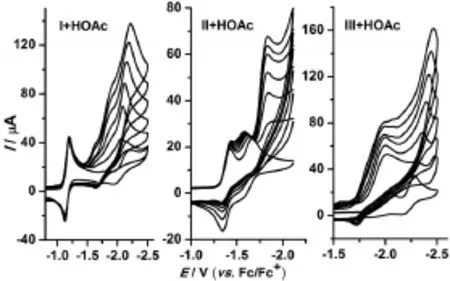
图4 模型配合物I、II、III(1 mM)催化HOAc还原产氢的电化学行为
图3给出了电催化质子还原过程中配合物I在-2.21V处、II在-1.72V处、III在-2.02、-2.28V处的电流强度随酸浓度增加的线性关系图,可以看出:电流强度与酸浓度均呈很好的线性关系。而直线的斜率说明了电催化还原峰对酸的灵敏度,即配合物III在-2.28V下有着较高电催化质子还原活性。

图5 模型配合物I、II、III在不同电位下电催化还原峰电流强度相对于乙酸浓度的变化
3 结论
本文合成了结构新颖的刚性共轭桥连[2Fe2S]模型配合物I,在对其进行PMe3配体取代研究的过程中,得到了单PMe3取代配合物II及单核铁双PMe3取代配合物III。吸电子共轭桥结构可有效地降低中心铁原子上的电子云密度,因此配合物I的第一还原电位(FeIFeI→FeIFe0)出现在-1.18V(vs. Fc/Fc+),成为目前具有最正第一还原电位的全羰基[2Fe2S]配合物。在乙酸存在的条件下,配合物I、II、III均能有效地电催化质子还原,其中配合物II在-2.28V(vs.Fc/Fc+)下有着最高的电催化活性。这些结果为小分子、低成本产氢体系的构筑提供了有价值的参考。
[1]杨秀云,张文娟,吴耀明,等.镁基储氢材料的研究进展[J].长春理工大学学报:自然科学版,2008,31(5):62-65.
[2]Lubitz W,Ogata H,Rüdiger O,et al.Hydrogenases[J].Chemical Reviews,2014,114(8):4081-4148.
[3]Tard C,Pickett C J.Structural and functional analogues of the active sites of the[Fe]-,[NiFe]-,and[FeFe]-hydrogenases[J].ChemicalReviews,2009,109(6):2245-2274.
[4]Frey M.Hydrogenases:Hydrogen-activating enzymes[J].ChemBioChem,2002,3(2-3):153-160.
[5]CammackR.Bioinorganicchemistry:hydrogenase sophistication[J].Nature,1999(397):214-215.
[6]Nicolet Y,Piras C,Legrand P,et al.Desulfovibrio desulfuricans iron hydrogenase:the structure shows unusual coordination to an active site Fe binuclear center[J].Structure,1999,7(1):13-23.
[7]王殿巍,王凯,刘旺,等.一种Re(I)配合物的合成及其光学氧传感性质研究[J].长春理工大学学报:自然科学版,2016,39(3):100-103.
[8]Rauchfuss T B.Diiron azadithiolates as models for the[FeFe]-hydrogenase active site and paradigm for the role of the second coordination sphere[J]. AccountsofChemicalResearch,2015,48(7):2107-2116.
[9]Denny J A,Darensbourg M Y.Metallodithiolates as ligands in coordination,bioinorganic,and organometallic chemistry[J].Chemical Reviews,2015,115(11):5248-5273.
[10]Artero V,Berggren G,Atta M,et al.From enzyme maturation to synthetic chemistry:the case of hydrogenases[J].AccountsofChemicalResearch,2015,48(8):2380-2387.
[11]Capon J F,Gloaguen F,Schollhammer P,et al. CatalysisoftheelectrochemicalH2evolutionby diironsubsitemodels[J].CoordinationChemistry Reviews,2005,249(15-16):1664-1676.
[12]高尚.铁铁氢化酶活性中心模型配合物的合成和性质研究[D].大连:大连理工大学,2008.
[13]Lyon E J,Georgakaki I P,Reibenspies J H,et al. Carbon monoxide and cyanide ligands in a classical organometallic complex model for Fe-only hydrogenase[J].Angewandte Chemie,1999,38(21):3178-3180.
[14]Li Ping,Wang Mei,He Chengjiang,et al.Influence of tertiary phosphanes on the coordination configurations and electrochemical properties of iron hydrogenasemodelcomplexes:crystalstructuresof[(μ-S2C3H6)Fe2(CO)6-nLn](L=PMe2Ph,n=1,2;PPh3,P(OEt)3,n=1)[J].European Journal of Inorganic Chemistry,2005,(12):2506-2513.
[15]Zhao Xuan,Georgakaki I P,Miller M L,et al.Catalysis of H2/D2scrambling and other H/D exchange processes by[Fe]-hydrogenase model complexes[J].InorganicChemistry,2002,41(15):3917-3928.
[16]Felton G A N,Vannucci A K,Chen J,et al.Hydrogen generation from weak acid:electrochemical and computational studies of a diiron hydrogenase mimic[J].Journal of the American Chemical Society,2007,129(41):12521-12530.
Synthesis and Electro-catalytic Hydrogen Evolution of Iron(I)/(II)Complexes as Functional Models for the Active Site of[FeFe]Hydrogenases
GAO Shang,ZHANG Weiyi,YANG Tingting
(School of Materials Science and Engineering,Changchun University of Science and Technology,Changchun 130022)
In order to tune the reduction potential of[2Fe2S]model for the active site of[FeFe]hydrogenases,complex[μ-SC4N2H2S-μ]Fe2(CO)6(I)with the conjugated and electron withdrawing bridge was synthetized by the reactionofFe3(CO)12andpyrazine-2,3-dithiolintetrahydrofuranatrefluxingtemperature.Thediironcomplex[μ-SC4N2H2S-μ]Fe2(CO)5(PMe3)(II)and a monometallic complex[μ-SC4N2H2S-μ]Fe(CO)2(PMe3)2(III)were obtained in the PMe3ligand replacements.All the complexes were fully characterized by IR,1H,31P,13C NMR and mass spectroscopy.The IR analysis data suggested that the electron withdrawing bridge,pyrazine,led to decreased electron density at the iron centers,which was consistent with the electrochemical studies.The cyclic voltammograms indicated that complex I exhibited a low primary reduction potential at−1.18V vs.Fc/Fc+with a 270mV positive shift related to that of the benzene-1,2-dithiolate(bdt)bridged analogue[(μ-bdt)Fe2(CO)6].Under the weak acid conditions,all the complexes could electrochemically catalyze the proton reduction.The mononuclear ferrous complex III showed relatively higher activity during the formation of hydrogen,confirming its potential as a catalyst for hydrogen production.
hydrogenase;chemical mimic;cyclic voltammogram;electrocatalysis
O614.81+1
A
1672-9870(2017)01-0081-04
2016-09-26
国家自然科学基金(21201022);教育部高等学校博士学科点专项科研基金(20122216120001)
高尚(1981-),男,博士,副教授,E-mail:custgaoshang@126.com
