渔药药效学专题讲座
——第一章 渔药药物效应动力学基础(3)
2017-03-20汪建国
汪建国
(中国科学院水生生物研究所 研究员 博导)
渔药药效学专题讲座
——第一章 渔药药物效应动力学基础(3)
汪建国
(中国科学院水生生物研究所 研究员 博导)
(二)药物-受体学说
1.占领学说 C1ark于1926年提出的占领学说认为:药物效果与药物占领受体数目成正比,药物占领受体数目取决于细胞表面受体的密度和受体周围的药物浓度,当全部受体被占领,即达到Emax。占领学说只适用于激动药,但不能解释2个激动药激动同样受体而产生不同的Emax。
1954年Ariens修正了占领学说,他把决定药物与受体结合时产生效应的能力称为内在活性。药物与受体结合不仅需要亲和力,而且还需要内在活性才能激动受体而产生效应。只有亲和力而没有内在活性的药物,虽可与受体结合,但不能激动受体故不产生效应。
1956年Stephenson又提出,药物只占领小部分受体即可产生最大效应,未经占领的受体称为储备受体。因此,当不可逆性结合或其它原因而丧失一部分受体时,并不会立即影响最大效应。进一步研究发现,内在活性不同的同类药物产生同等强度效应时,所占领受体的数目并不相等。激动药占领的受体必须达到一定阈值后才开始出现效应。当达到阈值后被占领的受体数目增多时,激动效应随之增强。阈值以下被占领的受体称为沉默受体。
2.速率学说 1961年Paton 提出了受体速率学说。该学说认为影响药物效应的大小的主要因素不是受体被占领数量的多少,而是药物分子与受体的结合速率。认为每当一个药物分子和受体相碰撞时即可产生一定量的刺激,并能被传到相应效应器而产生效应。使用这一学说的简单之处在于,仅以受体-药物结合和解离速率参数即可推算结果,无须使用内在活性和效能等参数。但问题在于这一学说无法解释药物与受体多种类型的相互作用。故这一学说被使用的范围并不大。
3.二态模型学说 Kar1in A.和Changenx JP.分别提出了药物受体作用的两态学说,又称变构学说。它们都认为受体存在活性状态(R*)和非活性状态(R)。两者均可与药物结合,而且活性和非活性受体之间也可以相互转化,二者处于动态平衡。激动剂主要与R*结合形成R*A,而拮抗剂则主要形成RB。实际上激动剂和拮抗剂分别推动R*和R的转换向着各自方向移动。这两种状态的受体之比例及两种状态受体与药物的亲和力大小决定着生物效应的大小。这种模型考虑到了受体与药物间相互作用导致的受体活性改变,更接近于实际的药物-受体反应状况,但用以直接说明药物与效应之间的定量关系尚不完备。
4.诱导契合学说 Kosh1and等提出的诱导契合学说,可以较好地解释配基与受体结合的实际过程。即药物与受体蛋白结合时,可诱使受体的空间构象发生可逆改变,而且这种结构变化即可导致生物效应。而且这种学说也可以解释药物与受体之间的协作效应。

图7 N2型乙酰胆碱受体阳离子通道分子结构示意图
需要注意的是,上述这些受体学说大多只能在某种特定的条件或范围内解释受体与药物的作用和受体与效应之间的关系,均存在一定的局限性。
(三)受体的类型、分布与特性
药物与受体结合形成复合物,引起一系列细胞反应,从而引起最终效应。不同的受体所处位置、本身结构以及信息传导过程各有不同,所引发的最终效应也都不一样。
1.离子通道受体 存在于快速反应细胞膜上的受体,多为含离子通道的受体。离子通道受体的主要特征是:受体蛋白本身组成一个跨膜的离子通道(图7),通道的开或关控制一些离子的跨膜流量,并通过改变细胞内离子浓度影响细胞功能。通道的开关则由配基与受体的结合或解离控制。例如神经肌肉接头处的N2胆碱受体是由5个亚基在细胞膜内呈五边形排列围成的钠离子通道,当乙酰胆碱与受体结合后,钠通道开放,钠离子内流增加,膜去极化,肌肉开始收缩。
2.G蛋白偶联受体,间接影响离子通道或第二信使 这是目前已发现的种类最多的受体类型,其激动剂种类包括生物胺、蛋白激素、多肽激素、脑/肠多肽、花生四烯酸系列的活性物质,淋巴细胞活性因子等。这类受体位于细胞膜,都由一条肽链形成,其N-末端在细胞膜外,为配体的结合部位;C末端在细胞内,为G蛋白结合部位。而且肽链形成7个跨膜螺旋结构和相应的三个细胞外和三个细胞内环,所以这类受体又称为7跨膜区受体(7TM)。7TM受体不仅具有共同的结构特征,而且具有共同的细胞内信号放大方式。这种结构能够稳定G蛋白结构,使它们能被脂质双分子层紧紧固定并保证它们不能进入到胞质中去(图8)。
G蛋白是鸟苷酸结合调节蛋白的简称,存在于细胞膜内侧,由α、β、γ三个亚单位组成的三聚体,静息状态时与GDP结合。当受体与配体结合后,相应受体被激活,GDP-α、β、γ复合物在Mg2+参与下,结合的GDP与胞浆中GTP发生交换,GTP-α与β、γ分离并与相应的效应机制结合,同时配体与受体分离。α亚单位内在的GTP酶促使GTP水解为GDP,激活效应机制,从而恢复原来的静息状态。G蛋白主要有两类,一类为兴奋性G-蛋白(GS),激活腺苷酸环化酶(AC)使cAMP增加;另一类为抑制性G-蛋白(Gi),抑制AC,使cAMP减少。G-蛋白还介导心钠素及NO对鸟苷酸环化酶(GC)的激活作用。此外,G-蛋白对磷脂酶C、磷脂酶A2,Ca2+、K+离子通道等有重要调节作用。一个受体可激活多个G-蛋白,一个G-蛋白可以转导多个信号给效应器(effector),从而调节神经、心肌、平滑肌等多种细胞的功能。
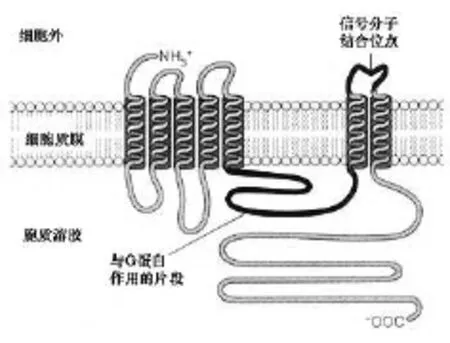
图8 G蛋白偶联受体的结构
3.跨膜激酶活性受体,直接调节蛋白磷酸化 这一家族包括许多多肽激素和生长因子的受体,例如上皮生长因子、血小板生长因子及某些淋巴因子等。它们位于细胞膜,只有一个疏水的跨膜区段,包括细胞膜外结合域、跨膜区、细胞内酶活性区几个部分。受体分子本身不具有激酶活性,当激动剂与细胞膜外的识别部位结合后,其细胞内某些可溶性酪氨酸激酶被激活,首先在特定的部位发生自身磷酸化,然后再将磷酸根转移到其效应器上,使效应器蛋白的酪氨酸残基被磷酸化,从而改变效应器的活性(图9)。

图9 跨膜激酶活性受体作用机制
4.核受体,对DNA的转录进行调节大多数甾体激素类受体属于这一类型,如性腺激素受体(雄激素受体、雌激素受体、孕激素受体)、肾上腺皮质激素受体(糖皮质激素受体、盐皮质激素受体)、甲状腺激素受体、维甲酸受体、维生素D3受体等。这类受体位于细胞内,主要是在细胞核上,细胞浆中也能检测到该类受体。这类受体和核内特定的DNA结合,并通过这种结合影响遗传信息的转录。当其被激活后,可通过对DNA转录的调节影响蛋白质的合成而导致生理或生化反应。例如肾上腺皮质激素激动受体所产生的抗炎作用就是这种机制。作用于细胞内受体的内源性配基或外源性配基都必须透过细胞膜后才能起作用,因此要求配基具有一定的脂溶性(图10)。
(四)受体与药物的结合
1.亲和力与内在活性 药物与受体结合形成复合物而引起效应,效应强弱遵循质量作用定律,即药物作用强弱与药物占领受体数目多少成正比,可用下列公式表示:
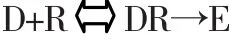
式中D代表药物,R代表受体,DR代表药物受体复合物,E代表效应。药物与受体结合后产生生理效应,不仅需要有亲和力,而且还需要有内在活性。亲和力是指药物与受体结合的能力,它反映出药物的作用强度(效价强度);内在活性是药物与受体结合后激动受体产生效应的能力,它反映出药物的最大效应(效能)。当亲和力相等,药物最大效应取决于内在活性;当内在活性相等时,药物作用强度取决于亲和力的大小。亲和力一般受采用药物-受体复合物的解离常数(KD)的倒数(1/KD)来表示,KD是引起最大效应一半时(即50%受体被占领)的药物剂量。
2.作用于受体的药物分类 根据药物与受体间亲和力与内在活性的不同,将药物总体分为激动药和拮抗药两大类
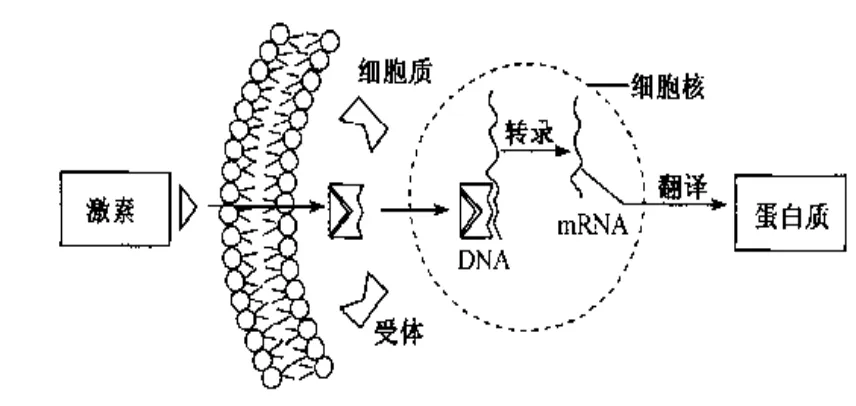
图10 核受体作用机制
(1)激动药 对受体既有亲和力又有内在活性的药物,能与受体结合并产生效应。根据亲和力和内在活性不同,激动药可分为:
①完全激动药,是具有较强亲和力和内在活性(α=1)的药物;
②部分激动药,是对受体有较强的亲和力,但内在活性较弱(0<α<1)的药物,单独应用,有较弱的效应,可与激动药竞争受体;
③负性激动药,也称反转激动药,激动受体后产生激动药相反的效应。负性激动药与受体结合后使受体构型发生质的改变,引起与激动药相反的效应。
(2)拮抗药 也称阻断药,是对受体有较强亲和力,而无内在活性(α=1)的药物。根据药物与受体的结合的可逆性,拮抗药可分为:
①竞争性拮抗药,由于与受体的结合是可逆的,可与激动药竞争与受体结合;
②非竞争性拮抗药,由于与受体的结合是牢固和相对不可逆的,或结合后引起受体构型改变和活性降低,使激动药无法完全对抗这种非竞争性改变,因而效应降低。
3.受体与药物的相互作用
(1)竞争性拮抗 竞争性拮抗发生在:①阈剂量增加;激动药与拮抗药之间;②激动药与部分激动药之间;③拮抗药或部分激动药与内源性激动剂(递质、激素、内在活性物质)之间。由于激动药、拮抗药和部分激动药对受体都具有亲和力,可相互竞争同一受体,发生竞争性结合或拮抗。它们与受体的结合是可逆的,合并用药的效应取决于各自药物的浓度和亲和力。观察拮抗药或部分激动药对激动药量-效曲线的影响是研究竞争性拮抗的常用方法。通常,先做激动药的量-效曲线,把激动药从受体部位清洗干净后,加入固定浓度的拮抗药或部分激动药,再观察在拮抗药或部分激动药存在下激动药的量效曲线的变化(图11)。
激动药与拮抗药的竞争性拮抗激动药有强的内在活性,与受体结合可引起药理效应;拮抗药无内在活性,与受体结合后不产生效应。拮抗药存在时,激动药量效曲线发生下列变化:
②曲线平行右移(斜率不变),KD值增加;

图11 不同药量的竞争性拮抗剂、非竞争性拮抗剂、激动剂和部分激动剂相互作用量效关系图
③Emax不变。根据拮抗药对激动药量效曲线的影响,可测定拮抗药的pA2值,或是使激动药的KD值增加一倍时拮抗药摩尔浓度的负对数。pA2是使激动药浓度提高一倍时所产生的效应与原来的效应相同时所用拮抗药摩尔浓度的负对数,是衡量拮抗药亲和力的指标。pA2越大,拮抗药亲和力越强,对激动药的阻断作用越明显。
激动药与部分激动药的竞争性拮抗部分激动药存在时使激动药的量效曲线发生下列变化:
①阈剂量减小;
②在激动药剂量较小时,量效曲线左移,说明部分激动药起激动药的作用;
③在激动药剂量较大时,量效曲线右移,说明部分激动药起拮抗药的作用;④Emax不变。
(2)非竞争性拮抗 有些拮抗药与受体结合牢固,形成的复合物解离速度很慢,以致造成不可逆性结合,此时,增大激动药的浓度也无法达到原来的Emax。
非竞争性拮抗可使激动药的量效曲线发生下列变化:
①曲线非平行右移;
②Emax降低.根据非竞争性拮抗药使最大效应降低的程度,可测得其pD2′值。pD2′是指使激动药最大效应降低一半所需非竞争性拮抗药的摩尔浓度的负对数,是衡量非竞争性拮抗药亲和力的参数。
(五)受体的调节
受体虽是遗传获得的固有蛋白,但并不是固定不变的,而是经常处于代谢转换的动态平衡中,受体的数目、与配体的亲和力以及效应经常受内环境变化以及药物的影响而发生改变。各种因素使细胞上受体发生质和量变化的过程称为受体调节。受体调节的生理意义在于通过调节使机体能更好地适应内外环境的变化,例如受体反应性减弱可保护细胞免受过量或长期刺激而导致生理功能紊乱,但受体调节过度则又会引起一系列病理性的后果。
1.受体失敏 在与配基作用一段时间后,受体对配基的敏感性和反应性下降的现象称为失敏。失敏可以由于受体数目的减少和/或由于受体与配基亲和力降低所致,它通常具有剂量依赖性、时间依赖性和可逆性等特点。
失敏又可分为以下两种情况:失敏如果发生在与配基特异性结合的受体上,称为同种失敏,即某种受体被配基激活后引起的失敏仅为该受体本身,而同一细胞上的其他受体系统并无实质性变化。
失敏如果发生于配基的非特异的受体上,即某种激动剂在作用一段时间后,不仅使其特异性受体对其反应降低,还使同一细胞上其他受体对它们各自激动剂的反应减弱。例如长期在与去甲肾上腺素接触后,细胞不 仅对去甲肾上腺素的反应性降低(同种失敏),而且对前列腺素的反应性也降低(异种失敏)。
2.受体增敏 增敏是与失敏相反的一种现象,即当配基与受体作用一段时间后,受体的数目增加和/或亲和力增加的现象。增敏也可发生于同种受体或异种受体,例如长期使用β肾上腺素受体阻断剂的情况下突然停药可出现受体敏感性比正常增高(同种增敏);又如大鼠长期饲以甲状腺素后可出现心肌中β肾上腺素受体的合成增加,结合位点增加(异种增敏)。
(未完待续)
(通联:430072,中国科学院水生生物研究所 武汉市武昌东湖南路7号)
