磷酸钒钠Na3V2(PO4)3电化学储能研究进展
2017-03-13宋维鑫侯红帅纪效波
宋维鑫 侯红帅 纪效波,*
(1中南大学化学化工学院,长沙410083;2Department of Materials,Imperial College London,London SW72AZ,UK)
磷酸钒钠Na3V2(PO4)3电化学储能研究进展
宋维鑫1,2侯红帅1纪效波1,*
(1中南大学化学化工学院,长沙410083;2Department of Materials,Imperial College London,London SW72AZ,UK)
锂离子电池在全球范围内的广泛应用加剧了对锂资源的消耗,其成本和原料将限制其未来发展。钠与锂具有相似物理化学性质,并且储量丰富。根据锂离子“摇椅式”电池原理,富钠离子化合物可类似富锂离子正极材料,提供可脱嵌的钠离子及结构。钠离子较锂离子大,其可逆脱嵌反应要求材料结构具有较大的容钠位与离子迁移通道。聚阴离子体磷酸钒钠Na3V2(PO4)3属于钠离子超导体(NASICON)材料,其NASICON结构骨架形成了稳定的容钠位,并且开放的三维离子迁移通道利于提高钠离子的扩散。Na3V2(PO4)3作为电池正极材料,具有理想的比容量、电压平台与循环稳定性,从而受到了广泛关注。本文首先介绍了Na3V2(PO4)3结构特点,其次结合团队已有的工作基础对Na3V2(PO4)3在钠离子电池、混合离子电池、水系电池,混合超级电容器等体系中的应用与反应机理进行了阐述;总结了基于Na3V2(PO4)3设计的复合材料与结构并探讨了Na3V2(PO4)3可能存在的问题与未来发展趋势。
Na3V2(PO4)3;钠离子超导体;电化学;能源存储;材料结构
1 引言
电化学储能相比于机械储能、电磁储能和相变储能具有效率高、成本低、安全方便等特点,已发展成为当前主要的储能技术。传统的铅酸、镍镉电池等电化学储能体系容易产生重金属污染,镍氢电池成本高,限制了其广泛应用。锂离子电池是一种在能量密度和功率密度上均占有优势的蓄电池,可用于电子产品,航空航天,军事军工等众多领域。随着锂离子电池的广泛应用,尤其是电动汽车市场的快速发展,锂资源被大量消耗并将濒临枯竭1-4。当前的锂离子电池技术在规模储能阶段存在着价格高、材料稳定性差、长循环安全性能差等问题5,6。同时,综合锂离子电池材料制造、电池生产和循环的能耗考虑(图1),制造1 kWh的锂离子电池需要耗能约400 kWh,产生约75 kg的二氧化碳气体(相当于35 L汽油燃烧放出的气体量),其主要能耗为电极材料的生产。因此,锂离子电池只有循环工作数百次(>400)后,其环境效益才能逐渐显现。

图1 制造1 kWh锂离子(Li-ion),与镍氢(Ni-MH),铅酸(Pb-acid)电池所需能耗比较1Fig.1 Energy cost for the production of 1 kWh battery comparing among Li-ion,Ni-MH,and Pb-acid batteries1
锂元素地壳含量仅0.0065%,全球锂储量76%以上集中分布在南美洲。锂资源储量少,全球分布不均,使得锂离子电池成本一直居高不下。同主族的钠元素地壳中含量约为2.8%,丰富度为锂资源的430倍,并且与锂具有相似的物理化学性质(表1)7-10。根据锂离子“摇椅式”电池原理,富钠离子化合物可类似富锂离子正极材料,提供可脱嵌的钠离子及结构,匹配对应的电解液、隔膜与负极构成室温钠离子电池7,11-13。钠离子电池的发展则为降低二次离子电池成本提供了重要方向10,14-16。钠离子电池通过钠离子嵌入电池正负极电势的不同产生电池电压,并实现钠离子在正负极之间的嵌入和迁出,完成电荷储存和释放。
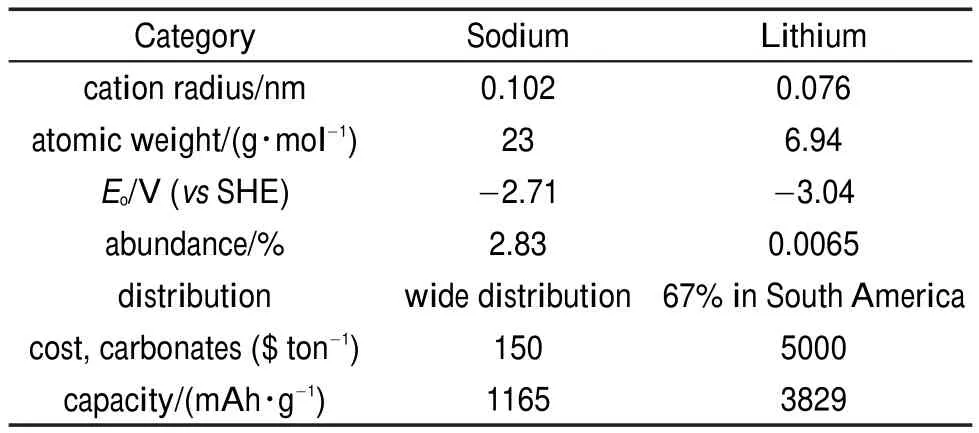
表1 钠与锂的物理化学性质比较Table 1 Physiochemical characteristics of sodium and lithium

宋维鑫,1990年生。2012年、2015年分别获中南大学工学学士、硕士学位。2015年获英国帝国 理 工 学 院 (Imperial College Lodon)校长奖学金于材料系攻读博士学位。主要研究方向为基于功能化石墨烯的复合材料设计与电化学研究,能源存储和转化。

侯红帅,1988年生,2016年博士毕业于中南大学化学化工学院,获“湖南省优秀毕业生”,主要研究方向为锂/钠离子电池电极材料。现为中南大学化学化工学院副教授。

纪效波,1980年生。2007年博士毕业于牛津大学,现为中南大学化学化工学院教授、博士生导师,英国皇家化学会会士,国家优秀青年基金获得者。主要研究方向为新能源材料与能源器件,主持国家自然科学基金4项。
钠离子电池近年来已成为热点研究问题,各类储钠材料已被广泛研究用作二次钠离子电池电极材料12,13,17-24。图2展示了可储存钠离子的正负极材料及其理论容量和电压关系13。正极材料主要包括层状材料、聚阴离子体材料、高分子聚合物材料等,负极材料包括相转换型材料(硬碳,钛酸钠等)、合金类材料(锡,锑等)等。钠离子超导体(NASICON)具有特殊的骨架结构,是快速钠离子导体,如Na3Zr2PSi2O12,主要组成形式为AxMM′(XO4),其中A为碱金属元素,MM′为Fe、Ti、Sc、V、Zr等具有3d能级的金属元素,X一般为W、P、S、Si、Mo25。Na3M2(PO4)3(M=Ti,Fe,V)10,11,26-31,Na3V2(PO4)2F332-35等聚合物体系具有开放式三维NASICON结构,适合钠离子快捷迁移,可提高具有较大半径和重量的钠离子在电化学迁移过程中的动力学特性,因而是一类理想的钠离子电池正极体系。Na3V2(PO4)3(NVP)相比于其它过渡金属NASICON结构材料具有易制备、大容量、高电位特点。NVP电化学反应过程V4+/V3+和V3+/V2+氧化还原电对分别产生3.4和1.6 V(vs Na/Na+)的工作电压,理论容量可达176 mAh·g-1,离子扩散系数为~10-11cm2·s-1。当前,NVP作为电极材料已被应用于钠离子电池、混合离子电池、水系电池、混合超级电容器等体系,并具有理想的电化学储能特性。

图2 钠离子脱嵌电极材料理论容量与工作电压区间关系13Fig.2 Theoretical capacity and working voltage of Na-insertion/de-insertion electrode materials13
本文首先介绍了NVP结构特点,总结了基于NVP设计的复合材料与结构体系,并结合团队已有的工作基础对NVP在不同电化学储能体系中的应用与反应机理进行了阐述,并探讨了NVP可能存在的问题与未来发展趋势。
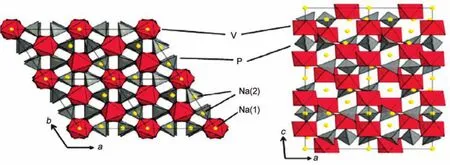
图3 菱形NVP不同视角下的结构示意图36Fig.3 Crystal structure at different orientations of rhombohedra NVP36NVP:Na3V2(PO4)3
2 Na3V2(PO4)3结构
2.1 六方晶族γ-NVP
NASICON结构材料由“灯笼式”骨架单元组成,每个结构基元包含三个XO4四面体和两个MO6八面体并通过公用角相连,形成可以容钠碱金属离子的两类离子占据位M(1)和M(2)。M(1)和M(2)可能被离子全占据或部分占据。菱形NASICON结构的NVP结构模拟图如图3所示,每个晶胞包括六个NVP结构基元。每个结构基元由八面体VO6和四面体PO4通过多面体角相连,成为聚阴离子体[V2(PO4)3],并在c轴方向通过PO4与相同聚阴离子体相连。NVP具有高度开放的三维结构,能产生很大的间隙空间供给钠离子迁移37。主要有两类不同氧环境的钠离子位于该晶体的空隙或通道中,其中一类是六配位环境位于八面体位置的容钠位(6b),定义为Na(1)位,每个式单元包含1个Na(1)位;另一类是八配位环境位于四面体位置的容钠位(18e),定义为Na(2)位,每个式单元包含3个Na (2)位28,38。因此,一个结构基元中的聚阴离子体[V2(PO4)3]最多可容纳四个单价碱金属离子。但在合成过程中,由于二价V2+极不稳定,从而NaxV2(PO4)3中的钒价态一般为三价V3+,而控制电化学反应在一定电压区间内进行则可以实现钒价态的转变36。
Delmas等40在1978年最早通过高温固相反应,利用NaPO3与V2O3在500和800°C下分别煅烧15 h,制备出了空间群为R3¯c,菱形结构的NVP,晶胞参数为a=0.867 nm,c=2.171 nm。经过高温反应合成的NVP具有有序排布的晶体结构,空间群为R3¯c的六方晶族被作为NVP的经典体系。直到1992年,有关NVP的深入研究才被Gopalakrishnan等39报道。他们将Na2CO3、(NH4)H2PO4和V2O5进行混合后,在氢气氛围下高温煅烧24 h制得NVP。NVP的晶胞参数(表2)与原子坐标信息(表3)也同时被总结,现已收录至SpringerMaterials数据库。Gopalakrishnan等同时指出,Br2与CHCl3作为氧化剂通过回流法可以逐步移除NVP中的钠离子,得到相应的去离子体系Na2V2(PO4)3和NaV2(PO4)3。但是通过X射线衍射图谱进行分析,表明NaV2(PO4)3非常不稳定而发生了分解,因此只有Na2V2(PO4)3体系的晶胞参数与原子坐标(表3)得到了总结。Gopalakrishnan等进一步通过氯气在CHCl3中氧化NVP,移除了NVP中的全部钠离子并得到新的V2(PO4)3物相。V2(PO4)3具有与NVP相同的晶体结构,但其晶胞参数(表2)与原子坐标(表3)发生较小变化。V2(PO4)3作为可插NASICON体系,其腾空的嵌钠位能够重新被锂离子、氢离子等阳离子插入而成为新的NASICON体系。

表2 Na3V2(PO4)3,Na2V2(PO4)3和V2(PO4)3晶胞参数与空间群(SG)39Table 2 Cell parameters of Na3V2(PO4)3,Na2V2(PO4)3,and V2(PO4)3with space group(SG)39
Goodenough等41在2001年合成了NVP,并通过离子交换法在锂盐溶液中制备了Li2NaV2(PO4)3。他们指出在NVP结构的NASICON体系中,相比于Na(1)→Na(2)→Na(1)形式的离子传导机理,离子传导更趋向于以Na(2)→Na(2)形式进行。根据六方晶系模型计算出的晶胞参数为a=0.8642(9) nm,c=2.172(3)nm,V=1.405(5)nm3。2010年,Zatovsky42针对NVP结构进一步展开了研究,指出六配位氧环境的位Na(1)位上钠的原子占据位为0.805(18),八配位氧环境Na(2)位上钠的原子占据位为0.731(7)。Zatovsky同时指出NVP属于三方晶系(关于NVP晶系分类将在后文总结),晶胞参数为a=0.87288(2)nm,c=2.18042(7)nm,V=1.43873(7) nm3。

表3 Na3V2(PO4)3,Na2V2(PO4)3,V2(PO4)3原子坐标39Table 3 Atomic coordinates of Na3V2(PO4)3,Na2V2(PO4)3and V2(PO4)339
2012年,Lim等43通过密度泛函理论计算了NVP结构,指出如果结构基元NVP中所有的Na(1)位和Na(2)位都被钠离子占据,则会形成具有四个钠离子的Na4V2(PO4)3体系,一个钠离子占据Na(1)位,三个钠离子占据Na(2)位。NVP晶体结构中一个钠离子占据Na(1)位,占据位为1,两个钠离子占据Na(2)位,占据位为0.67。同时,他们利用热重-差式扫描量热技术发现在450°C以下的温度范围内,NVP和去钠NaV2(PO4)3体系都不会发生任何相变反应。原位加热XRD也表明,从25°C升温至450°C过程中,NVP和去钠NaV2(PO4)3体系都没有产生新的二次相。NVP在450°C以下的温度范围内能够保持较高的热稳定性,其结构中稳定的P―O化合键对于提升此类材料热稳定性具有重要作用。2014年,Jian等44采用强氧化剂NO2BF4移除NVP中的钠离子,得到了室温下稳定的物相NaV2(PO4)3。他们进一步采用Rietveld精修确定了室温下NVP和NaV2(PO4)3的晶胞参数与原子坐标。NVP中Na(1)位原子占据位为0.843,Na(2)位占据位为0.719。NaV2(PO4)3中Na(1)位原子占据位为0.9448。通过像差校正环形明场扫描透射电子显微技术(ABFSTEM)和核磁共振(NMR)技术对比分析了NVP和NaV2(PO4)3中的容钠位,证实了NVP中以Na(2)→Na(2)形式进行的主要离子传导过程。他们同时研究了不同温度下,钠离子占据位的变化。当温度降低至100 K时,NVP中Na(1)位和Na(2)位原子占据位分别变为1和0.667,说明钠离子在低温时趋向占据Na(1)位使整个NVP体系在低温下达到新的热力学平衡状态。
2015年,Masquelier等45研究了不同温度下NVP的晶体结构,并在200°C下通过同步加速器X射线衍射技术进行材料表征,指出NVP具有六方晶系结构。经过Reitveld结构精修得到NVP中Na(1)位(Wyckoff位为6b)和Na(2)位(Wyckoff位为18e)的原子占据位分别为0.75(3)和0.70(4)。根据他们的原子占据位分布,每个NVP晶胞中含有17.1个钠原子。虽然他们在文章中表示该数值与Z=6的化学式单元结果近似一致,但与Z=6的式单元18个钠原子这一结论具有一定差别。NVP中钠位均被钠部分占据,因此都能够参与钠离子的传导。NVP中钠原子的非均质热因数与原子坐标信息,说明了Na(1)位上的原子在垂直于[001]的方向,Na(2)位原子在稍微偏离[001]的方向具有较大的迁移率。该结论说明了NVP与其他NASICON体系存在的经典M(2)→M(1)→M(2)的离子迁移模型一致。图4为γ-NVP沿[001]方向的结构示意图,在每个“灯笼式”结构两端分布着1个Na(1)位离子,并同时被6个Na(2)位离子包围。因此钠离子在Na(1)位和Na(2)位两类钠位上呈现无序排布。
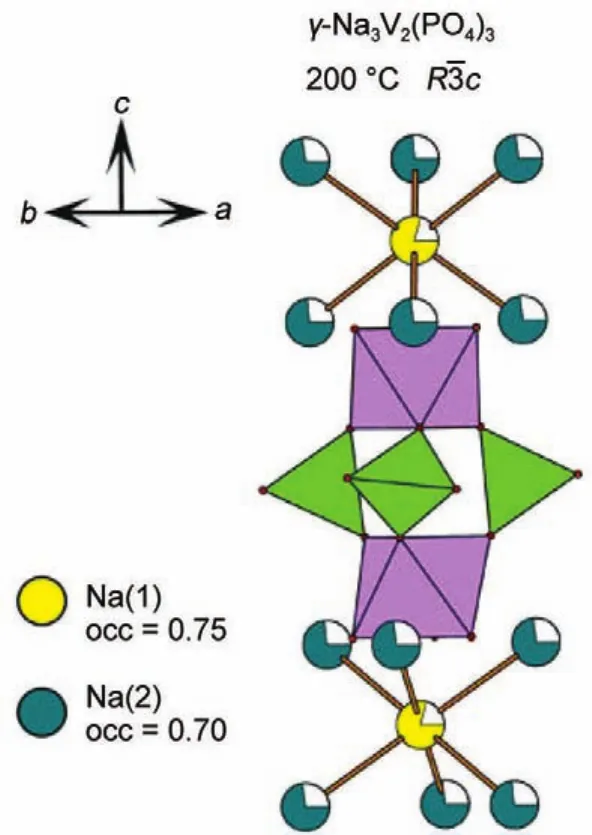
图4 γ-NVP沿[001]方向结构图,八面体为VO6,四面体为PO445Fig.4 Structure of γ-NVPalong[001]direction with VO6octahedra and PO4tetrahedra45occ:occupation
γ-NVP空间群一般认为R3¯c按照晶体结构点群进行归类的“七大晶系”理论,应属于三方晶系(trigonal),并且是菱方居中(rhombohedral-centering)的六方晶格。根据γ-NVP晶胞参数特点,γ-NVP属于布拉维六方晶系(Bravais hexagonal)。由于布拉维晶系中包括了菱方,晶胞参数只能满足a=b=c,α=β=γ≠90°的关系。因此对六方晶族的总结,γ-NVP属于六方晶族(crystal family),或归为三方晶系,或归为布拉维六方晶系(见表4)。
2.2 单斜晶系α-NVP
尽管大多数关于NVP的研究都表明NVP是具有菱形对称结构,属于R3¯c空间群的快钠离子导体,但Masquelier等45-47多次报道了低温下具有单斜特点超晶胞结构NVP。2000年,Masquelier与Nazar等47通过高分辨同步加速XRD进行研究,表明NVP大部分强衍射峰分裂成2-3个属于单斜对称的特征峰。计算得到的晶胞参数为a=1.5124 nm,b=0.8733 nm,c=0.8847 nm,β=124.65°。其它低强度的衍射峰来自于室温下钠离子有序排布所产生的超结构。2014年,Masquelier等45再次通过400和700°C两段高温固相反应合成NVP并对其进行研究。图5为实验所得XRD图及精修结果,说明NVP属于C2c空间群的单斜晶系,其晶胞参数分别为a=1.5112 nm,b=0.8727 nm,c= 0.8824 nm,β=124.54°。同时,他们指出,当温度高于200°C时,具有单斜对称特点的NVP会转化为菱形对称的钠离子无序排布结构,即R3¯c。2015年,Masquelier等46率先利用差示扫描量热技术(DSC)研究了NVP在-30-225°C范围内的相变,并总结出升温过程中三个可逆相变过程:25.8°C发生α→β相变,118.6°C发生β→β′相变,177.2°C发生β′→γ相变。冷却过程三个相变温度与升温过程的相变温度相比,发生不同程度的偏移,如图6所示。α与γ相发生相变时具有类似尖锐的热效应峰,而γ相是被认为具有R3¯c空间群的菱形对称结构,α相对应的晶体结构则一直未被研究。
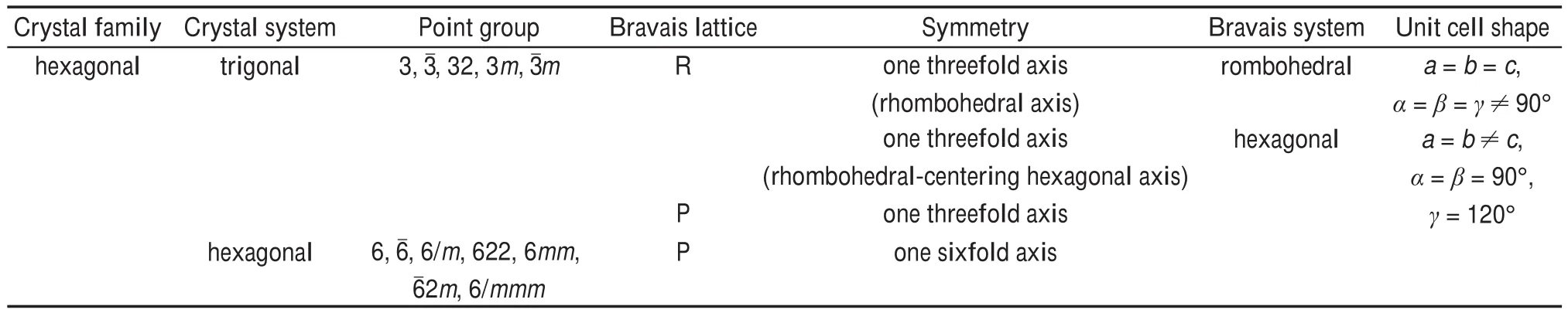
表4 六方晶族与布拉维晶系关系Table 4 Hexagonal crystal family with related Bravais crystal system.

图5NVP在室温25°C的XRD图谱及其精修结果46Fig.5 XRD pattern of NVPat 25°C with refinement46
Masquelier等45利用低温XRD衍射技术通过实验光源对-10°C时的NVP进行了研究,得到了使用同步加速光源11-BM和实验光源进行衍射得到的NVP在不同温度下的XRD图。在测试温度降低至-10°C时,NVP逐渐呈现了单斜对称特点。NVP在20°C时XRD中出现超结构。-10°C测试下NVP在Q=0.088 nm-1出现了一个新的衍射峰,低温下NVP衍射峰在γ-NVP菱形对称(116)和(211)位置发生了分裂。-10°C下NVP的单晶衍射如图7所示,图谱中出现的超结构衍射斑与γ-NVP菱形对称关系不相符。经过计算,低温时出现的超结构峰属于C2c空间群的单斜晶系。α-NVP与γ-NVP之间的晶胞参数符合以下矩阵转换关系:
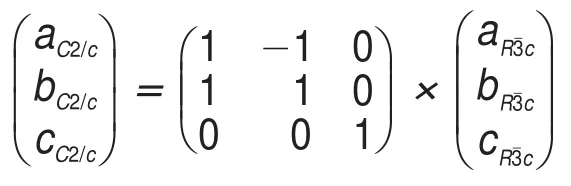

图6 NVP在-30-225°C范围内的差示扫描量热曲线及其对应的不同晶体结构45Fig.6 DSC curve of NVPfrom-30 to 225°C with four different crystal structures(α,β,β′,γ)45

图7 NVP的(0kl)层在-10°C时单晶X射线衍射图谱45Fig.7 Precession image from single crystal X-ray diffraction of the NVP(0kl)layer measured at-10°C45
α-NVP晶胞含有Z=12个NVP结构基元,其晶胞体积是γ-NVP的2倍。α-NVP晶胞中含有3个V,5个P,5个Na和18个O,其原子坐标信息列于表5中。根据NASICON结构中由八面体和四面体构成的“灯笼式”结构模型,α-NVP对称性的降低说明在结构中出现了更多独立的Wyckoff位。γ-NVP中Na(1)位分裂为α-NVP中Na(1a)(4a)和Na (1b)(8f),Na(2)位分裂为Na(2a)(8f),Na(2b)(8f)和Na(2c)(8f)。精修后钠原子占据位为0.97-1.05,结构单元为Na2.94V2(PO4)3,因此可以认为α-NVP中所存在的Na位被钠离子完全占据,原子占据位修订为1。根据该分配规则,每个α-NVP晶胞中会存在12个NVP式单元,包括12个Na(1)位和24个Na(2)位占据的钠离子。

表5 α-NVP原子坐标与钠位之间的非均质热因数(Uij,单位nm2)Table 5 Atom coordinates of α-NVPand anisotropic temperature factors(Uij,in nm2)of Na sites
α-NVP(C2c)因此可以被认为是钠无序排布的γ-NVP(R3¯c)相的有序相。如图8所示,α-NVP中Na (1)位上有序排布钠离子时,结构中同时会产生空位配体形成Na(1)[Na(2)4□2]八面体。α-NVP和γ-NVP相中Na(2)/□的有序排布如图9所示。在(ab)面上,Na(2)位离子的分布均呈现六边形组成的“蜂巢状”排布。γ-NVP相中钠离子无序排布在Na (2)位,六边形中心为部分占据Na(2)位的钠离子。α-NVP相中六边形中心为空位,会改变R3¯c中菱形对称形式。在c轴方向上,每六层Na(2)/□排布面按照一定向量关系偏移并沿c轴旋转60°即可满足单斜晶体周期性。六边形中Na(2)位之间的原子距离较大,钠离子在其中的扩散形式并未被研究说明。
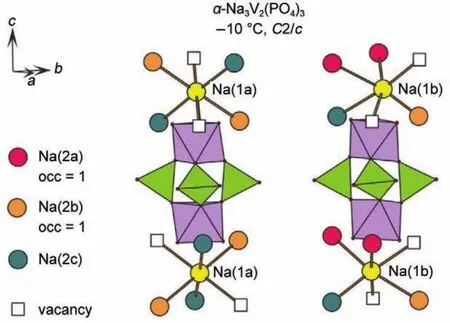
图8 α-NVP沿[001]方向结构图45Fig.8 Structure of α-NVPalong[001]direction45
2.3 β-NVP和β′-NVP
Masquelier等45通过DSC曲线发现在介于α-NVP(C2c)和γ-NVP(R3¯c)相之间,存在两类过渡相,分别为β-NVP(30-10°C)和β′-NVP(125-170°C)。NVP相变位于β-NVP和β′-NVP之间时,会出现不属于菱形对称的超结构衍射峰。NVP从高温(200°C)冷却至20°C时,(116)和(211)位置的衍射峰发生了明显的分裂。尽管他们之前报道过该类超结构衍射峰属于单斜对称,但同步加速XRD中出现的超结构衍射峰并不完全符合单斜对称。结合同步加速XRD图谱和单晶衍射图谱,β-NVP和β′-NVP精修后的晶胞参数列于表6中。Masquelier等指出β-NVP和β′-NVP可以认为是经过不对称调整后的单斜结构,也可能是α-NVP和γ-NVP相之间不完全转换而形成的过渡相结构。

图9 α-NVP(a)和γ-NVP(b)中Na(2)位离子分布结构示意图45Fig.9 Projection in the(ab)plane of the Na(2)arrangement in(a)α-NVPand in(b)γ-NVP45
3 NVP电化学储能机理
3.1 钠离子电池
NVP钠离子电池在电压区间2-4.6 V(vs Na/ Na+)内,循环伏安曲线中出现的氧化峰(Na离子脱出)和还原峰(Na离子嵌入)分别位于3.7和3 V,且有一对不明显峰出现在2.3 V左右,完成两个离子的脱嵌38,48。原位XRD、ICP、K-edge XANES等测试说明了两个钠离子在充电过程中脱出NVP形成物相NaV2(PO4)3的过程49-51。非原位的XPS测试通过测定NVP中钒V3+经充电氧化变为V4+再经放电还原为V3+的过程,说明存在两个钠离子的嵌入和迁出反应实现钒价态之间的转变6。随着循环次数的增加,发现还原峰位置不断正移,而氧化峰位置基本没有出现偏移,使氧化还原峰电位差不断减小,说明NVP材料在钠离子嵌入和迁出的循环过程中会经过结构的重整优化33。
NVP/Na钠离子电池在2-4.6 V(vs Na/Na+)电压区间内,以0.1C,0.2C,0.5C和1C电流密度充放,克容量分别为117.6、111、104和100 mAh· g-152,其中1C表示两个钠离子在1 h内脱嵌单位NVP时的电流,不同电流密度下能够产生的电池。首次充放电库伦效率为94.4%、92%、92.8%和97%,即使在高倍率电流充放时也表现出较高的效率,主要是由于通过碳热还原法制备的NVP表面结构包覆碳,电导率得到提升。NVP钠离子电池极化电压仅为0.04 V,而Jian38和Cong53等在文献中报道过0.07 V的极化电压。
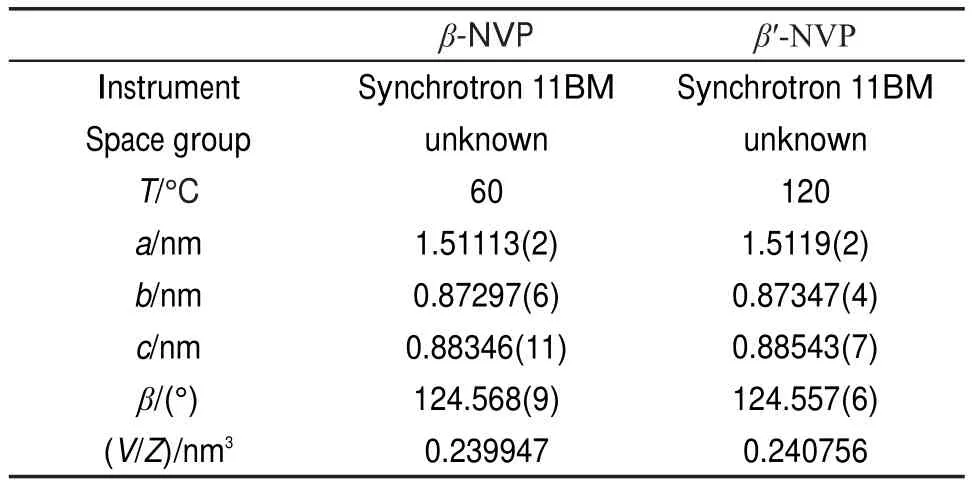
表6 XRD精修计算得到的β-NVP与β′-NVP晶胞参数45Table 6 Cell parameters of β-NVPand β′-NVPfrom XRD refinement45
NVP具有两类不同氧环境的钠位,Na(1)位处于六配位环境,在两个沿着z轴方向相邻的八面体V2(PO4)3结构所形成的空隙中,Na(2)位具有八配位环境,位于沿z轴方向相邻的四面体PO4结构中并与磷原子相平行的位置54。Houria28和Lim43等已经报道如果所有Na(1)位和Na(2)位被钠离子占据,则可形成Na4V2(PO4)3,其中Na(1)位容纳1个离子,Na(2)位容纳3个离子。NVP钠离子电池可以通过控制电压区间,实现钠离子嵌入和迁出,进行V4+/V3+和V3+/V2+之间的氧化还原反应55。由于V2+在Na4V2(PO4)3中相对不稳定,很难通过化学反应制备出含有四个钠离子的Na4V2(PO4)3材料,而是形成稳定V3+的NVP37,因此通过碳热还原法制备时,即使有过多的碳原料存在于反应物中,在600-800°C范围内最终也只是能够制备出V3+的NVP。钠离子电池充放电过程中,离子迁移个数将对电池比容量产生重要影响,而NASICON结构NVP不同钠位的占据位不同,对脱嵌离子数目会产生不同影响56,57。
为了确定NVP中不同钠位的离子占据位,并解释NVP脱钠过程,我们采用第一性原理进行计算时以表3中NVP和脱钠Na2V2(PO4)3的原子坐标信息进行建模。通过计算钠离子脱嵌引起的晶体结构参数变化,再以此来验证表中的结论。在第一性原理计算中所用超晶胞组成为[Na3V2(PO4)3]2。计算过程中的Na001,Na002,Na003,Na004,Na005,Na006表示Na(2)位中占据的钠离子,离子占据位为0.75;Na007,Na008表示Na(1)位中占据的钠离子,离子占据位为0.540。为了研究NVP中两类不同的钠位对所占据的钠离子迁移造成的影响,我们首先利用第一性原理对比分析了NVP中钠原子和氧原子之间的键布局及原子布局。通过对比数据可以发现,在Na(2)位上占据的钠离子与Na(1)位上的钠离子相比,具有较小的键布局,说明该位置周边的限制环境较弱,离子相对容易脱出晶体结构。因此在Na(2)位上占据的钠离子所具有的离子迁移行为将对NVP钠离子电池的电化学性能产生决定性作用,研究Na(2)位离子占据位变化也将能够阐释NVP中钠离子迁移过程。
Lim等43确定过NVP中钠离子的占据位,钠离子在Na(1)位上的占据位为1,在两个Na(2)位上的占据位为0.67。然而根据他们的结论,按照超晶胞[Na3V2(PO4)3]2组成(包括6个Na(2)位钠离子和2个Na(1)位钠离子),则在两个Na(2)位上占据的钠离子首先全部脱出时,相对于式单位NVP晶胞约为两个钠离子(计算结果实为2.01个钠离子)。虽然多余的0.01个钠离子产生的容量可以忽略,但是按照Yu等对钠离子占据位的分配,对应计算出未进行钠离子迁出时的单位化合物组成为Na3.01V2(PO4)3,而不是NaV2(PO4)3。按照Landolt-Börnstein Database in Springer Materials晶体数据库对NVP钠离子占据位的分配,则能成功解释式单位NVP两个Na(2)位脱出两个钠离子的过程,并在分析过程中保证原始物相组成与NVP相一致。
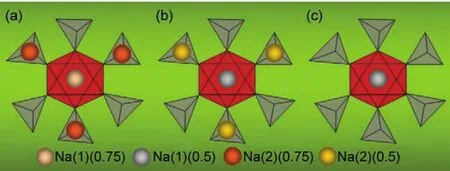
图10 [Na3V2(PO4)3]2计算单元离子占据位变化示意图54Fig.10 Scheme representing ions occupations based on the calculated[Na3V2(PO4)3]2unit model54
图10为[Na3V2(PO4)3]2计算单元在进行钠离子脱出过程中的离子占据位变化示意图。图10(a)是DFT计算模型中Na(1)位和Na(2)位上的离子占据位均为0.75的Na3V2(PO4)3原始晶胞。Na(1)位离子由于周围原子布局的影响迁出时需要较高的能量,而Na(1)位占据的离子位能相对较低,所以当Na(2)位离子存在于体系中时,会优先迁出。第一个钠离子从NVP晶胞中脱出时,[Na3V2(PO4)3]2中6个Na(2)位离子占据位会由0.75发生减小。与此同时,超晶胞中2个Na(1)位离子由于体系变化会发生晶体结构的重整,占据位由0.75发生减小。为了使Na(1)位与Na(2)位被离子占据时位能最低,超晶胞[Na3V2(PO4)3]2中迁出2个钠离子后,Na(1)位与Na(2)位原子占据位经结构优化平衡为0.5。
当体系中Na(1)位与Na(2)位离子占据位变化为0.5后,产生一个钠离子脱出的Na2V2(PO4)3晶胞,如图10(b)所示。由于体系稳定后,Na(1)位离子相比于Na(2)位离子具有较低的能量,因此再进行离子脱出时,Na(2)位上离子会首先脱出。根据超晶胞[Na2V2(PO4)3]2中钠离子占据位进行计算,在第二步迁出1个钠离子过程中,由于Na(2)位具有较弱的限制氧环境,会优先迁出体系。6个Na(2)位上占据位为0.5的钠离子,有4个Na(2)位中占据位为0.5的钠离子会变为0,2个Na(1)位上占据位为0.5的钠离子不发生变化。则相对于超晶胞[Na2V2(PO4)3]2是从Na(2)位中迁出了2个钠离子,钠脱出后的超晶胞[NaV2(PO4)3]2中6个Na(2)位上4个为空位,2个被钠离子占据,占据位为0.5;2个Na(1)位上被钠离子占据,占据位为0.5,达到如图10(c)所示的结构。此时,NVP则能够完成两个钠离子迁出并转化为脱钠结构NaV2(PO4)3,产生理论比容量117 mAh·g-144,55。NVP钠离子电池在0.2 mV·s-1扫速下的循环伏安测试,3.72和3.06 V处具有一对明显的氧化还原峰,对应V3+/V4+氧化还原反应54。曲线中同时出现了凸起,表示离子占据位发生变化时进行结构重整。
图11(a)表示了NVP中不同钠位的分布情况。图11(b)则是DFT计算过程中优化后的晶体结构所对应的XRD图,属于R3¯c空间群。NVP中位于Na (2)位上的离子标识为Na1、Na2、Na3、Na4、Na5、Na6,Na(1)位上的离子标识为 Na7、Na86,37,39。图11(c)表示NVP中Na(2)位钠离子脱出体系,成为NaV2(PO4)3的过程。当6个Na(2)位上离子完全脱出,其占据位会由0.75到0.5再变为0,完成两个钠离子迁出NVP并达到NaV2(PO4)3。NaV2(PO4)3中Na(1)位上的离子键布局发生降低,O―Na键长变短,产生较强的诱导效应,致使离子迁出较难。该结论也说明了NVP转变为NaV2(PO4)3时,Na(1)位中钠离子将更难脱出体系,致使电化学充电过程需要更高电压平台下,以更高的能量使其发生氧化反应。同时,Na(2)位离子迁出后O―V键长变短,但钒键布局基本未变,主要由于离子迁出时钒发生氧化反应,使八面体VO6发生收缩,导致相应晶格参数的减小。DFT理论计算说明了钠离子迁出NVP后晶胞参数均都减小。
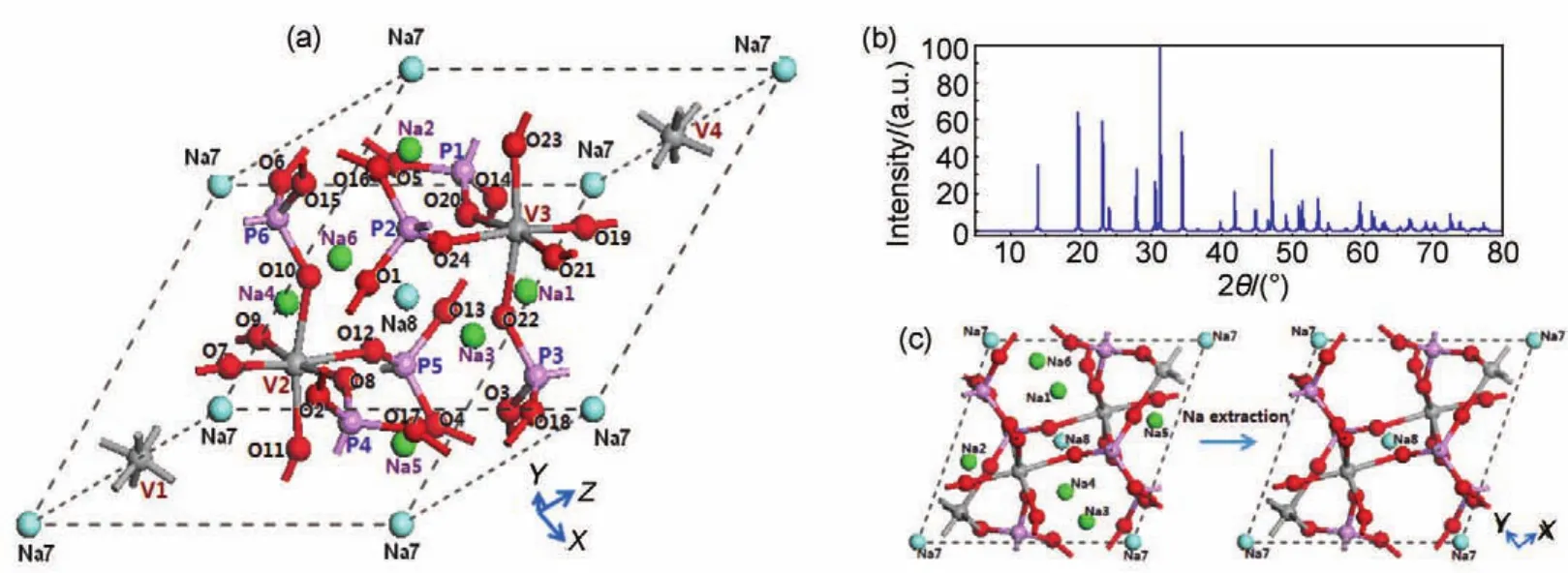
图11 Na3V2(PO4)3结构示意图53Fig.11 Schematic representation of a refined Na3V2(PO4)3(NVP)structure53
假设[Na3V2(PO4)3]2超晶胞中6个Na(2)位上的离子通过一步完成迁出,则相当于超晶胞中脱出了4.5个钠离子,实现NVP到Na0.75V2(PO4)3的转化。钠离子一步迁出后的晶体结构与NVP相比,轴长减小2%,晶胞体积减小10%。钒的总电荷数从3.21增加到了5.3。总电荷数非整数增大了2.09,说明NVP中三价钒V(III)被氧化为四五价混合价态V(IV/V),主要由于一步离子迁出过程中,大于2个钠离子参与了氧化反应。根据Faraday定律计算[Na3V2(PO4)3]2超晶胞中6个Na(2)位上的离子通过一步迁出,则相当于Na3V2(PO4)3中脱出2.25个钠离子,产生比容量为131 mAh·g-1。然而,大量的实验数据证明NVP在充电过程中不能够实现如此大的摩尔容量6,37,38,说明一步迁出钠离子的理论与实验数据也相违背。

图12 NVP电化学反应过程中分别沿(a)x、(b)y、(c)z方向的离子迁移路径54Fig.12 Possible Na ion migration paths in NVPalong(a)x,(b)y and(c)curved z directions54
经过第一性原理的计算,NVP中Na(2)位的离子会优先在电化学反应过程中进行嵌入和迁出52,54,但不同脱嵌路径所需活化能不同。对于三维NASICON结构的材料,在包括空位跃迁的相邻钠位之间可以考虑三条可能的迁移路径58。图12(a)为钠离子沿着x轴,在两个PO4四面体之间的通道进行迁移的路径。图12(b)为钠离子沿着y轴,在PO4四面体和VO6八面体之间的空间迁移的路径。图12(c)为钠离子通过八面体结构后,在经过PO4四面体和VO6八面体之间的空间经过的曲线路径。根据所示的几种迁移路径,钠离子按照设定的x、y、z路径进行离子迁移,可以应用第一性原理计算出各个路径下的迁移能量分别为0.0904 eV、0.11774 eV和>200 eV。通过迁移能的数值,说明了NVP中钠离子的迁移可以较容易地沿x、y两条主要的路径进行离子迁移,而不能沿着z轴直线方向进行离子迁移。当钠离子需要在z轴方向进行迁移时,在DFT计算过程设定了一条如图13(c)中所示的曲线路径进行,并对其所需的迁移能进行计算。当钠离子沿曲线路径绕过VO6八面体结构,在进入PO4四面体和VO6八面体之间的间隙,所需要的活化能为2.438 eV,相比于z轴直线方向迁移的活化能已经大大降低,因此是一条在z轴方向上可行的离子迁移路径。因此,通过DFT理论计算说明了钠离子在NVP结构内进行迁移运动时,可在三维方向上具有快速迁移的路径,而最可能的两条路径则是沿x与y轴直线方向。
第一性原理计算说明NVP三维NASICON结构能够为钠离子在三维方向上提供具有快速迁移的路径,对NVP钠离子电池中离子扩散系数产生重要影响。NVP钠离子电池以0.1、0.2、0.5、0.8、1 mV·s-1不同扫描速率进行循环伏安测试得到峰电流-扫描速率关系可以看出,氧化还原峰电位差随着扫描速率增大而不断增加,说明了在大电流密度下电极的不可逆性也在不断增大。同时,通过峰电流ip和扫速二分之一次方(v1/2)之间的线性关系,可以判断出NVP钠离子电池的电化学反应过程属于扩散控制过程。同时,该体系电池所产生的钠离子扩散系数(DNa+)可以根据Randles-Sevcik公式34,36,59-61进行计算。

其中m为正极活性材料质量,F为法拉第常数,R为气体常数,T为绝对温度,n是氧化还原反应中转移电子数,因为在实验设定的反应区间内能够实现两个离子的脱嵌,因此n=2,A为电极和电解液之间接触的有效面积,通常以电极片的几何面积代替近似60,A=0.78 cm2,D则是需要计算的扩撒系数,C是晶体结构中可进行迁移的钠离子浓度,根据NVP晶胞体积可以计算出C=2.35×10-6mol·L-3。根据氧化还原峰电流计算得到NVP在钠离子电池中的扩散系数分别为1.05×10-11和1.03× 10-11cm2·s-1。NVP在钠离子电池中的扩散系数与在锂离子电池中的相比36,具有较低的扩散速度,主要原因是与锂离子(或混合离子)电池相比,在钠离子电池中是具有较大体积和重量的钠离子进行离子迁移,完成充放电过程中的氧化还原反应。同时,通过比较NVP在钠离子电池和锂离子电池中的扩散系数,也说明了在混合离子电池体系中,是以锂离子为主的迁移过程,从而观察到在混合离子电池中表现出较高的离子扩散系数。
3.2 混合离子电池(锂离子电池体系)
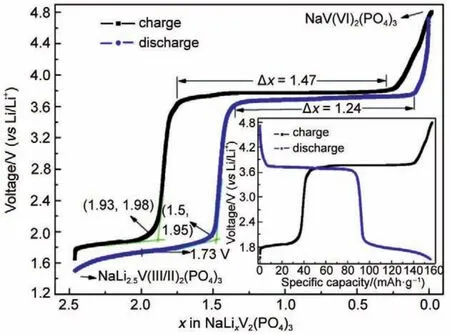
图13 静置过24 h的混合离子电池电化学电压组成曲线和0.1C下首次充放电曲线图36Fig.13 Electrochemical voltage-composition curves of the first galvanostatic cycle at a current rate of 0.1C for 24 h stated NVPhybrid-ion battery36
通过使用钠离子正极材料替代锂离子正极材料应用于锂离子电池体系中,可设计发展一类具有钠、锂混合离子迁移效应的新型混合离子电池体系,将对二次电池多元化发展产生重要意义。同时,研究富钠离子正极在混合离子体系中的离子迁移机理,能够为开发设计高效、低成本嵌锂正极材料,提高锂离子电池对正极材料中嵌入锂离子的利用率,以节约对锂资源的消耗,降低锂离子电池成本。
NVP混合离子电池在0.5 mV·s-1下进行循环伏安测试,具有明显的氧化还原峰62。氧化还原峰主要是由于两个钠、锂混合离子嵌入和迁出一个NVP单元体系,相应发生V4+/V3+转化。同时,首次循环伏安曲线与随后的曲线发生了较大差别,其氧化还原峰位置逐渐发生偏移,并且其中一对氧化还原峰(氧化峰~4.4 V(vs Li/Li+),还原峰~3.4 V(vs Li/Li+)几乎随循环次数的增多而逐渐消失。在首次还原曲线中表现出了两个较强的还原峰,表示钠锂共同嵌入正极体系时,锂离子凭借其较小的半径和质量易进行嵌入,而此时钠离子也会发生一定程度的嵌入,最后相对于一个晶胞结构,完成两个或大于两个离子的嵌入。在随后的循环测试中,由于钠离子的不可逆性以及半电池负极提供的过量锂离子,导致逐步变为以锂离子为主的迁移,钠离子的嵌入和迁出也逐步弱化从而导致一对越来越弱的氧化还原峰。同时,从曲线的峰位置可以发现,随着循环次数增加,其氧化还原峰差值也在逐渐增大,说明了不断增大的电极极化致使电化学反应的可逆性也逐步变差33。
NVP混合离子电池在分别经过1、10、24、72 h室温常压条件下静置后,在0.1C电流密度下1.6-4.6 V(vs Li/Li+)充放电区间内,电池克容量与电压平台产生了差异36。对于静置1、10 h的混合离子电池,首次放电容量相近,约为127 mAh· g-1,充电平台约在3.77 V,放电平台约在3.65 V。对于静置24、72 h的混合离子电池,首次放电容量相近,约为148 mAh·g-1,充电平台约在3.76 V,放电平台约在3.68 V。同时,混合离子电池静置时间达到24 h以后,与未静置到24 h的电池相比,其平均氧化还原电压平台差由0.12 V变为0.08 V,说明经过充分静置后,电池极化电压会降低。单位NVP经过非原位XPS测试说明在两个钠离子脱出和嵌入过程中,可实现两个钒V3+发生氧化还原反应与V4+进行转化7,则以此计算出的理论容量为117 mAh·g-1。对于实验过程中,NVP混合离子电池产生的大容量和较小的电极极化,主要原因是由于电解液和正极材料之间发生了较充分的离子交换,使具有更轻、更小半径的锂离子为主要碱金属离子在磷酸钒钠中发生嵌入和迁出。而对于富钠离子正极材料与富锂电解液之间的离子交换原理,已经通过ICP-AES进行了研究与确定51。嵌入的锂离子替代了钠离子占据NVP结构中的Na(2)位,由于锂离子迁移相比于钠离子具有更快的速度和更好的可逆性,因此在循环过程中离子的嵌入和迁出也会逐步以锂离子为主。发生离子交换的电极体系,由于其他离子的掺入所表现出的比容量会超过NVP的理论容量。根据Faraday定律(C=26.8n/w,其中C表示理论容量,n是单位反应中的电子转移数目,w是反应活性物质质量分数),比容量将因w的减小而变大。
NVP混合离子电池在0.1C下充放电曲线,第六次充放电曲线与首次相比,在反应的电压平台处更加稳定,在其他电压位置则更加陡直,说明混合离子电池与开始仅有钠离子进行迁移的电池体系而言,具有了更快的离子嵌入和迁出速度。经过24 h静置之后的混合离子电池能够在不同倍率下,具有更大的容量和更好的循环稳定性。
经过24 h静置的NVP混合离子电池,在0.1C倍率下进行首次充放电的电压组成曲线如图13所示,插图中为测试过程中放电容量为148 mAh·g-1的首次充放电曲线。电压组成曲线是假设首次充放电都是由钠离子进行迁移完成时,由实验测试的混合离子充放数据,计算出相对于单位磷酸钒钠晶胞进行嵌入和迁出的钠离子个数。实际在电化学反应的嵌入和迁出过程中,锂离子是在嵌入/脱出过程中的主要离子,在电压组成曲线中以锂离子数目代表混合离子脱嵌过程。由于相对于标准氢电极电势,锂电对Li/Li+和钠电对之间相差0.3 V,根据磷酸钒钠在钠离子电池中V4+/V3+(3.4 V vs Na/Na+)和V3+/V2+(1.6 V vs Na/Na+)的电压平台,推算实现NVP锂离子电池中V4+/V3+和V3+/V2+转换的平台约在3.7 V和1.9 V(vs Li/Li+)。由于有混合离子电池中会有部分钠离子嵌入负极作为正极的对电极,因此负极电极电势相对于锂片负极稍高,对应电压平台又会比3.7和1.9 V(vs Li/Li+)稍低。
根据测试曲线可以计算,对于在3.7 V左右实现V4+/V3+氧化还原反应的充放电平台,充电平台中有1.47个离子脱出,放电平台有1.27个离子发生嵌入。对于首次循环过程电压平台处迁出和嵌入离子数目的差异,主要是因为迁移离子性质变化引起的。离子嵌入(放电)过程中,由于钠离子具有较大半径和较重的质量,钠离子迁移具有较大的不可逆性,因此嵌入离子中锂离子含量会大大提高。同时,可能在负极端形成的SEI膜,能允许锂离子顺利通过但对钠离子的通过和传导产生了一定的阻碍作用,使得嵌入过程中的钠离子含量减小。根据Faraday定律可知,当该电位平台下的理论容量一定时,锂离子不断嵌入体系时形成的物相M会因钠离子被锂离子替换(假设全部为钠离子时嵌入量与假设全部为锂离子时嵌入量相同)而减小,因此嵌入的离子个数(主要以锂离子为主的混合离子体系)会减小。钠离子在脱出体系时需要更多的能量,而相同数目的锂离子因为更小、更轻则只需要较少的能量就能够嵌入正极体系。同时,可以通过分析离子迁移的极限情况来说明混合离子迁移过程。当首次放电至设定电压为1.6 V时,体系中嵌入碱金属离子数目为2.5个。假设嵌入离子全部为锂离子,达到物相NaLi2.5V2(PO4)3时按照Faraday定律计算可以得到所产生的放电比容量为156 mAh·g-1,远高于实验中测试的实验值148 mAh·g-1,说明在嵌入过程中除了不可逆因素造成钠离子的不可逆外,仍有部分钠离子参与了离子迁移,使表现出的放电容量发生减小。
实现V4+/V3+氧化还原反应,需要有两个钠离子进行脱嵌并达到117 mAh·g-1的理论容量。假设嵌入过程全为锂离子实现两个锂离子嵌入达到NaLi2V2(PO4)3,则产生126 mAh·g-1的比容量,相当于放电电压为1.73 V处。假设嵌入过程全部为钠离子实现两个钠离子嵌入则产生117 mAh·g-1的比容量,相当于图14中放电电压为1.76 V处。由于混合离子的嵌入过程,在第一个平台结束时能够嵌入1.5个离子,另外0.5个离子完成嵌入则需一直持续放电至1.73-1.76 V之间。深度放电到截止电压1.6 V,会使额外0.5个离子继续嵌入,进行V3+/ V2+还原反应,使体系中的钒达到V(II/III)的混合价态。


图14NVP菱形NASICON结构发生离子交换示意图37Fig.14 Idealized representation of the rhombohedral NVPNASICON structure37
图14为NVP结构与离子交换示意图,阐释了NVP的内部容钠位置以及在混合离子电池体系中的离子交换过程。每个NVP结构基元有3个Na,分别占据着2个Na(2)位和1个Na(1)位。如果NVP的3个Na(2)位可被充分占据,则可形成容纳最多4个Na的Na4V2(PO4)3。但由于该物象中V的价态不稳定,导致Na4V2(PO4)3无法在实验过程中直接合成。对此,Goodenough等41表示该材料中Na(1)位的钠离子是不能活动进行离子交换的,只有Na(2)位上占据的离子才能够进行离子交换和迁移。但同时Nazar47和Gopalakrishnan39等也表示单位磷酸钒钠中的三个钠离子都是可以完全脱出的,而关于该问题的争议可能是由于磷酸钒钠材料制备方法不同造成的。
根据Nazar47和Gopalakrishnan39等的理论,可以用公式(2)表示,其中□表示Na(2)位的空位,Na+1(2)表示Na(2)位的第一个钠离子,Na+2(2)表示Na(2)位的第二个钠离子,Na+(1)表示Na(1)位的一个钠离子。公式(2)中的离子迁移机理表示三个钠离子均可以进行离子交换,而在嵌入过程由于钠离子的不可逆因素势必会造成锂离子主导的嵌入,类似锂离子向V2(PO4)3嵌入的电化学过程,然而却与本实验数据不相符。同时,如果Na(1)位的离子可以脱出并能进行离子交换,但是Goodenough等41在实验过程中研究了NaLi2V2(PO4)3经过100次充放电循环后,会始终存在一个钠离子在正极体系中,也与公式(2)中表示的机理相冲突。因此,本实验过程中将Na(1)位的离子确定为不可进行迁移的离子,主要由Na(2)位的离子参与离子交换和电化学反应过程。
如图14所示,NVP中Na(2)位占据的钠离子,会在静置过程中与电解液中的锂离子发生离子交换,而Na(1)位的钠离子则不易进行离子交换。NVP的Na(2)位存在一个空位,但在合成NVP时不会被Na占据。通过控制一定的电压区间,进行电化学嵌入后,使Na(2)位至多容纳3个Na。在混合离子电池体系中,NVP材料晶格的两个Na(2)位上占据的Na,会在静置、电化学循环过程中发生离子交换作用,逐步实现NVP向NaLi2V2(PO4)3的转化。公式(3)和公式(4)则分别表示Na(2)位钠离子可以与锂离子进行离子交换,两处Na(2)位上的离子(钠离子或锂离子)也可进行相互之间的离子交换。在混合离子的嵌入过程中,第三个Na(2)位则能够被嵌入的Li所占据。由此可以根据法拉第定律,计算其产生的理论容量与嵌入离子数目之间的关系,推算出相应物象状态下的钒的价态。
通过以上对混合离子电池体系离子迁移机理的理解,随电化学循环次数的增加,迁移离子将主要变为锂离子并实现物相逐步转变为NaLi2V2(PO4)3。由于循环伏安测试中说明了该电极的扩散控制特点,因此在混合离子电池体系的循环充放电过程中,锂离子主导的混合离子迁移行为,会对电池的电化学性能起到主要作用。结合Randles-Secvik公式计算出NVP在混合离子体系中氧化反应和还原反应过程中的离子扩散系数D分别为2.42×10-10和1.54×10-10cm2·s-1。NVP混合离子电池体系计算出的离子扩散系数与Li3V2(PO4)3体系的离子扩散系数(10-8cm2·s-1)相比,相对较小,说明了在混合离子迁移过程中,仍然存在着体积和重量较大的钠离子,增加了离子迁移过程中的不可逆因素,从而导致了较小的离子扩散系数。
混合离子电池体系在组装完成静置过程中,以及NVP电极将逐步在电化学循环过程中,NVP会与富锂电解液发生离子交换并在充放电循环过程中逐渐由混合离子的迁移,变为以锂离子为主的迁移,在正极侧逐步由富钠离子的NVP转变为NaLi2V2(PO4)3。
混合离子电池体系NVP静置发生离子交换转变为NaLi2V2(PO4)3的过程如公式(5)所示,Na(2)位上的钠离子均可以被电解液中的锂离子替代,其中γ代表Na(2)位的空位。在电化学反应过程及充分静置产生的离子交换过程,也能够实现在Na(2)位上已占据离子之间继续进行离子交换,如公式(6)所示。同时,通过控制电池测试电压,可以影响嵌入正极体系的离子个数,如公式(7)所示。在设定的电压区间范围内,相对于一个Na3V2(PO4)3单元可以有多于2个离子进行嵌入,嵌入离子在分别占据了2个原被占据的Na(2)位之后,剩余离子则会嵌入Na(2)位原有的空位处,但该处离子也只能经过电化学反应过程进行离子迁移,而不能够在静置过程中与任何来源的离子发生离子交换作用。否则,将有三个锂离子存在的NaLi3V2(PO4)3化合物会经过电池静置而形成,但静置过程无法提供给锂离子嵌入到Na(2)位空位所需的能量。
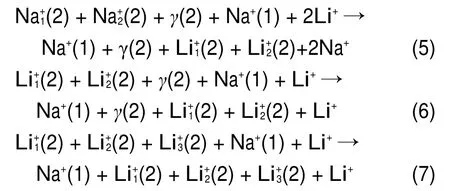
图15为NVP混合离子电池在0.1C倍率下,1.6-4.8 V的充放电区间内首次充放电的电压组成图。充电过程电压平台约为3.78 V,相对于NVP结构基元,能够有1.7个离子迁出。放电电压平台约为3.7 V,能有1.4个离子进行嵌入。极化电势约为0.08 V,说明通过S-CTR法制备的Na3V2(PO4)3具有良好的导电性,产生了较好的动力学特性。嵌入离子相比脱出离子数目有所减少,可以归结为SEI膜电阻等引起的不可逆因素使体积和质量较大的钠离子在迁移过程中数量减少,以及由于锂离子的主导嵌入过程使新物相质量分数减少,在电压平台(即发生氧化还原反应)处所产生的容量一定的前提下,按照Faraday定律计算可知嵌入的锂离子数目应比钠离子数目少。
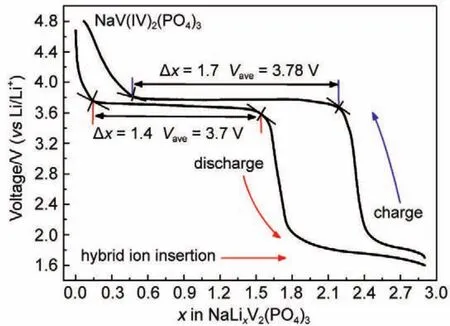
图15 NVP混合离子电池首次充放电压组成图62Fig.15 Electrochemical voltage -composition curves of the first galvanostatic cycle at a current rate of 0.1C for NVPhybrid-ion battery62
图15中的电压曲线根据实际测试容量曲线,按照只有钠离子进行脱嵌时产生的容量进行计算得到的曲线图。则当截止放电电压达到1.6 V,由于嵌入一个锂离子比嵌入一个钠离子产生的容量大,所以正极体系中能够至多嵌入2.9个锂离子,并实现四价钒到三价钒,再到二价钒的转化。假设体系嵌入的2.9个离子全部为锂离子,则可产生181 mAh·g-1的克容量,远高于实验测试值。说明嵌入的离子也不仅仅是锂离子,而是钠、锂混合离子嵌入过程。嵌入反应中,由于钠离子较大的体积和重量,会对锂离子的嵌入产生阻碍效应,如图16中所示,会进一步降低混合离子电池的循环稳定性和倍率性能。同时,大体积与大重量的钠离子在离子迁移过程中,也会因对锂离子造成的阻碍效应在一定程度上降低电池离子扩散系数。实验数据证明,经过充分的电池静置即富锂离子电解液与NVP发生充分的离子交换后,电化学性能明显提升。相比较经过短时间静置的离子交换反应,钠离子对锂离子迁移的阻碍效应将在电化学循环过程中表现尤为明显。因此,对于混合离子电池,充分静置将有助于提升电池性能和电化学稳定性。
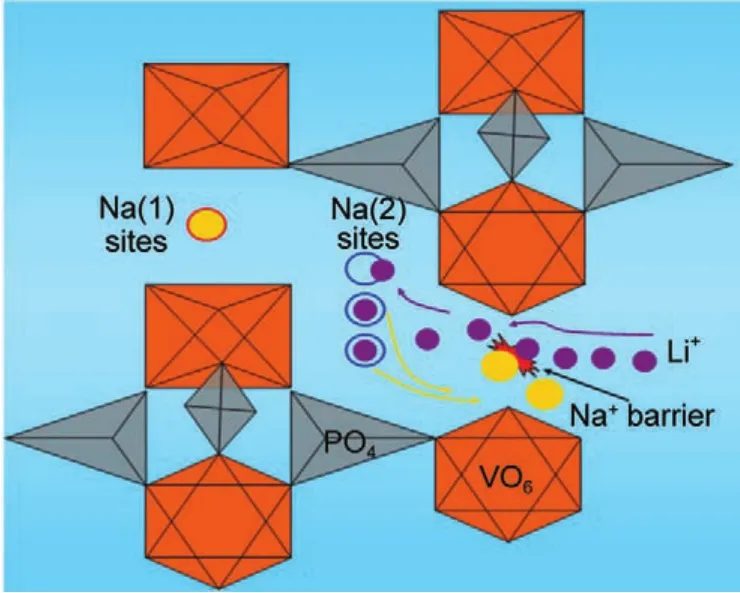
图16 Na3V2(PO4)3混合离子体系钠、锂离子迁移过程示意图62Fig.16 Representation of NVP-structure exhibiting the hybrid ion migration of Na and Li ions62

图17 NVP在Li2SO4,Na2SO4,K2SO4电解液中首次(实线)和第三次(虚线)循环伏安测试曲线63Fig.17 First(solid line)and third(dotted line)cyclic voltammograms(CV)curves of NVPin NVP在Li2SO4, Na2SO4,K2SO4electrolytes63
3.3 水系电池
NVP在1 mol·L-1的Li2SO4、Na2SO4、K2SO4电解液中,以5 mV·s-1的扫描速率在-0.2到0.9 V (vs SCE)电压区间内进行循环伏安测试,为了比较在中性电解液中NVP的电化学行为,其首次和第三次循环伏安曲线如图17所示。NVP在Li2SO4和K2SO4电解液中,一直需要经过循环伏安极化从而能达到稳定的反应过程,说明Na3V2(PO4)3电极在Li2SO4和K2SO4电解液中所具有的电化学反应活性相对较低64。图中非常不对称的氧化还原峰,说明了Li+和K+离子在NVP的电化学脱嵌过程中,电极过程呈现不可逆性。通过近似计算循环伏安曲线中氧化还原峰电位差,得到NVP在1 mol·L-1Li2SO4,Na2SO4和K2SO4中产生的峰差值分别为704、512和600 mV,说明了Li2SO4和K2SO4电解液中的Li+和K+离子在NVP的电化学反应过程中相对不可逆,具有较慢的反应动力学速度65。
NVP在Na2SO4电解液中在前三次循环伏安扫描过程中产生明显的对称氧化还原峰,说明Na+在NVP的电化学嵌入和迁出反应过程中,具有较优异的迁移特性。实验过程中所用的三类水系离子电解液,因为锂离子Li+(0.382 nm)和钾离子K+(0.331 nm)的水合离子半径分别大于和小于钠离子Na+(0.358 nm)水合半径66,同时钠离子与NVP结构体系具有更好的相容性,因而表现出了较优的电化学特性。如图18所示,水合离子半径中最大的水合锂离子Li+由于其较大的离子体积而很难在电化学反应过程中扩散进入NVP结构,且由于其离子传导率和离子迁移能力均不理想,因而在Li2SO4电解液中也很难产生电容储能行为66。由于K2SO4电解液中的钾离子K+具有最小的电子密度,与水分子之间能产生最弱的溶剂化能力,因而其具有最小的水合离子半径。钾离子K+与钠离子Na+和锂离子Li+相比,具有很高的离子传导率,因而会更加容易在NVP材料表面及孔结构表面形成双电层电容(EDLC),进行吸脱附反应而不会嵌入到晶体材料的内部结构中。Na2SO4水系电解液中的钠离子具有相对稳定的离子传导率和水合离子半径,获得一定程度双电层电容吸脱附反应的同时,主要会发生钠离子在NVP材料中的脱嵌反应,即伴有物相变化的电化学氧化还原反应,因而相比于钾离子水系电解液会产生更大的容量66。NVP在1 mol·L-1Na2SO4的水系离子电解液中以2、10、20、30 mV·s-1的高扫速进行循环伏安测试,均会产生一对明显的氧化还原峰。在高扫速的循环伏安测试中,快速充放电反应会在电极表面产生短时间内的暂态过程,使NVP利用最外层表面进行电荷储存。活性材料内部的孔结构会在较低速度扫描过程中用于电荷储存,并且伴随着离子的嵌入和迁出反应67。
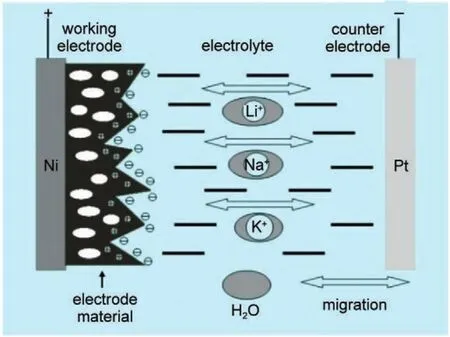
图18 NVP在Li2SO4,Na2SO4,K2SO4电解液中的电池和电容行为示意图63Fig.18 Battery and capacitance behaviors towards NVPelectrode in Li2SO4,Na2SO4,K2SO4electrolytes63
3.4 混合超级电容器
钠离子混合超级电容器是基于二次离子电池与超级电容器工作原理,由电池类可脱嵌离子型电极和双电层电容电极构成的兼有大功率和较高能量密度的新型储能器件。不同于非对称超级电容器组成是赝电容电极和双电层电容电极,混合离子超级电容器不适用赝电容电极,而通过发生氧化还原反应的电极进行离子脱嵌,能够进一步提高储能密度。Wang等68将NVP/C和活性炭作为混合离子超级电容器电极,在0-1.5 V电压区间内具有15.9 Wh·kg-1的能量密度。三电极体系中NVP,NVP/C和活性炭(AC)在0.5 mol·L-1Na2SO4电解液中不同电压区间内以5 mV·s-1进行循环伏安扫描,NVP和NVP/C于电压区间-0.2-1 V(vs SHE),AC于-1-0.4 V(vs SHE)内,分别产生179、197和120 F·g-1比电容。根据单电极电化学特性,可以确定两电极体系的工作电压区间,以同时实现两电极最大储能容量。两电极质量配比也需要根据单电极储能容量进行平衡,以提高器件的储能密度。
3.5 全固态钠离子电池
Masquelier等46将NVP分别作为正、负极电极材料,与固态电解质Na3Zr2Si2PO12(NZSP)组合成为“三明治”形式的全固态钠离子电池。正极、负极包括25%的NVP,60%的NZSP和15%的导电碳(CSP)。由于在充放电过程中,正极NVP会发生两个钠离子的脱嵌反应,实现与NaV2(PO4)3之间的相转换,而负极NVP发生一个钠离子的脱嵌反应,实现与Na4V2(PO4)3之间的相转换,因此为了平衡正负极容量,全固态钠离子电池正极材料(NVP/NZSP/CSP)为40 mg,而负极材料为80 mg,固态电解质NZSP为30 mg。全钠离子电池块体外观如图19左上所示,电池块体剖面组成如图19右上所示。电池在组装过程中,电极混合物和固态电解质分别在常温下压至50 MPa,再通过层层叠加方式组成电池并在900°C高温下煅烧10 min。得到的电池块体进行抛光并将两极与铂片相接,置于图中的高温电池内成为可以进行充放电的全固态钠离子电池。

图19 全固态钠离子电池块体外观图(左上),电池块体组成示意图(右上)和高温钠离子电池外形图(下)46Fig.19 Photo of the battery(top left),battery stack composition(top right)and design of the high temperature cell(down)46
在0-2.2 V区间NVP可发生氧化反应[Na3V2(PO4)3-2e-→NaV2(PO4)3+2Na+,V=3.4V(vs Na/Na+)],也可发生还原反应[Na3V2(PO4)3+Na++ e-→Na4V2(PO4)3,V=1.6 V(vs Na/Na+)]47。对称NVP全固态钠离子电池具有理论平台电压则为1.8 V。对比全固态钠离子电池在0.1C和0.5C下的充放电曲线,说明全固态钠离子电池在大电流密度下和充放电循环过程中会造成较大的容量损失和电极极化。极化增大的原因主要是由于NVP电极具有较大的颗粒并未进行碳包覆,其导电性较差。NZSP固态电解质也可能会在电化学反应过程中增大极化。电池在0.025C下仅前两个循环存在一定的不可逆容量,随后的充放电循环中表现出了非常优异的可逆性,但在0.5C电流密度下进行充放电循环,具有明显的容量损失。电池经过循环后,发现电极表面形成了较大的裂缝对离子和电子的传导产生影响。
Noguchi等69通过丝网印刷和热压技术制备了NVP并将其作为电极,利用NZSP固体电解质与金属钠首先设计了NVP/NZSP/Na全固态半电池。在充放电过程中伴随钠离子的脱嵌反应,也出现了3.4和1.6 V两个电压平台。随后,他们将NVP用作两极,工作电压区间设定为0.01-1.9 V,在1.2和10 μA·cm-2电流密度下,比容量为68和32 mAh·g-1。

图20 碳包覆纳米NVP在钠离子半电池体系中0.2C下(a)充放电曲线和(b)循环特性,碳包覆纳米NVP同时用作正负极的全电池体系中2C下(c)充放电曲线和(d)循环特性70Fig.20 (a)Charge/discharge profiles and(b)cyclic performance of nano NVP@C in sodium ion half cell at 0.2C in 1.3-2 V;(c)Charge/discharge profiles and(d)cyclic performance of Nano NVP@C as both cathode and anode in sodium ion full cell at 2C in 1-2.2 V70
3.6 NVP对称电极钠离子电池
对称电极电池能够降低电池成本,并且理论上可以不区分正负极进行充电71。Duan等70将碳包覆的纳米NVP用于钠离子半电池中,以0.2C倍率电流在1.3-2 V(vs Na/Na+)的电压区间内进行充放电测试,产生了70.7 mAh·g-1比容量,并在1.64 V处产生电压平台,对应于V3+/V2+氧化还原反应。该半电池体系经过50次充放电循环后,容量保持率为93.2%,如图20(a,b)。由于NVP可以通过V4+/ V3+之间的氧化还原反应产生3.4 V的电压平台,因此NVP既可以提供高电压平台,也可以提供低电压平台。Duan等将碳包覆的纳米NVP同时用作正、负电极设计了钠离子全电池,其中根据包覆的纳米NVP作为正、负极时产生的比容量确定全电池中正、负极质量比为3:5,电解液为1 mol·L-1NaClO4的碳酸丙烯酯溶液。该全电池开路电位接近0 V,正、负极在充放电过程中发生的氧化还原反应如下式所示:
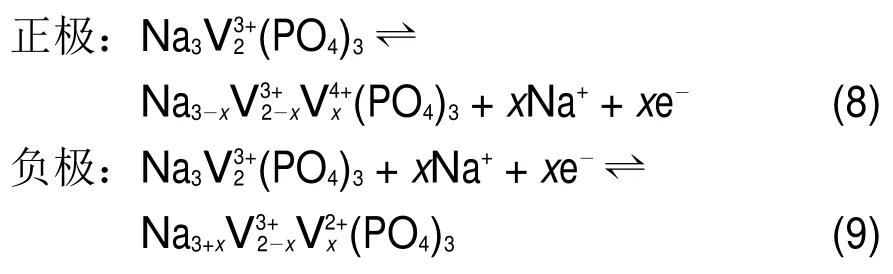
如图20(c,d),全电池在1-2 V电压区间内,分别产生一对充放电平台,在2C倍率的电流下,全电池中正极比容量可达90.9 mAh·g-1,其能量密度高达154.5 Wh·kg-1。经过50次充放电循环,容量保持率为81.4%。Plashnitsa等71也较早报道过NVP在钠离子全电池中的电化学特性,指出NVP对称全电池采用1 mol·L-1NaClO4的碳酸丙烯酯溶液做对称电池电解液,会发生电解液分解产生SEI膜并造成容量损失。NVP对称全电池电压最优区间为1.85 V。为此,他们采用0.4 mol·L-1NaBF4的EMIBF4离子液体作为电解液,正、负极质量比为1: 3,得到了分别为102和64 mAh·g-1的充放电容量。但相比于有机电解液,离子液体电解液室温下粘度大,离子传导性差并影响电极电解液界面,导致电池放电比容量低。Jian等72将电压区间设定在0-3 V研究了NVP在钠离子半电池中的电化学行为,发现在1.6 V和0.3 V存在电压平台,分别对应于钠离子嵌入NVP中的18e和6a位置形成Na4V2(PO4)3和Na5V2(PO4)3。两个钠离子嵌入NVP的过程可以分别产生约59和54 mAh·g-1的比容量。在0-3 V电压区间内,以1 mol·L-1NaPF6的碳酸乙烯酯和碳酸二甲酯(体积比1:1)溶液作为电解液,首次放电容量达277 mAh·g-1,可逆充电容量为149 mAh·g-1。An等51以1 mol·L-1LiPF6的碳酸乙烯酯和碳酸二甲酯(体积比1:1)溶液作为电解液,NVP为两极构建了对称混合离子电池。根据这一结构模型,非对称电极混合离子电池采用了NVP和钛酸锂(LTO)作为正负极。测试电流为100 mA·g-1,电压区间1-3 V内进行充放电,对称混合离子电池NVP-NVP具有79 mAh·g-1比容量,非对称混合离子电池NVP-LTO具有73 mAh·g-1比容量。
4 材料合成与储能特性
NVP具有NASICON开放式三维结构,V3d与O2p轨道能级相差大,从而导致其电导性差。NVP在碱金属离子脱嵌过程中产生的体积形变大,所产生的晶格应力会使颗粒表面产生缝隙而不稳定,造成容量的衰减及副反应的发生73。目前,通过减小颗粒尺寸(纳米化),表面提供包覆碳网络,体相掺杂与形貌控制等技术可以提高材料的导电特性,缩短离子和电子传输距离,提高其储能特性。
4.1 NVP/碳复合材料
4.1.1 无定型碳包覆
碳包覆是一类广泛用于碳热还原法以提高材料导电特性的方法,导电碳可在高温固相反应中原位包覆在材料颗粒表面形成导电网络,是一种固相合成电池电极材料使用最广泛、操作易控制、成本相对较低的制备方法。我们先后采用碳热还原法已成功合成了碳包覆的NVP36,52,62,74、Na3V2(PO4)2F333,34,54,75,76、Na2FePO4F59等富钠离子正极材料,并用作二次离子电池电极(包括钠离子电池和混合离子电池)。
通过比较NVP前驱体与NVP红外光谱,发现350°C煅烧的前驱体没有出现NVP特征峰。谱图中577和1050 cm-1处的峰表示PO4四面体中的P―O键作用,在629和978 cm-1处表示在八面体VO6结构中振动V3+―O2-键。1150-1250 cm-1之间的特征峰,表示PO4四面体的伸缩振动77。碳热还原反应法制备的NVP表面形貌无规则,颗粒尺寸为100-200 nm之间。固相合成材料颗粒在碳热反应过程中,由于碳源的存在而在高温煅烧中的生长受到限制,因此可保持较小的粒径。二次机械研磨会造成颗粒在煅烧过程中发生团聚。热重分析说明了碳热还原制备的NVP/C复合物碳含量为6.9%。
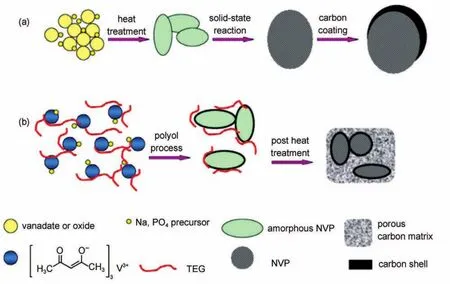
图21 (a)传统固相法合成碳包覆磷酸钒钠,(b)软化学法制备双层碳包裹的NVP78Fig.21 (a)Traditional solid-state way to prepare carbon-coated NVP,(b)soft-chemistry method for double carbon embedded NVP78
如图21所示,传统的碳包覆方法容易造成碳不均匀或不完全包覆颗粒表面,从而影响最终的性能。Zhu等78采用软化学法制备了具有多孔碳基体,镶嵌碳包覆NVP的新结构。软化学法应用前驱体原料分散在多元醇中,中温分解后得到均匀碳包覆的NVP,再将其与四甘醇混合进行二次煅烧得到双层碳包裹的NVP。四甘醇能够使预分解的颗粒分散,并在高温反应中作为还原剂。双层碳不仅可以提供导电网络,而且能有效抑制颗粒高温生长和团聚。在电流密度20C(1C电流密度为117 mA·g-1)下,双层碳包裹的NVP作为钠离子电池电极产生极小的电极极化,并且仍具有100 mAh·g-1比容量。在200C大倍率下充放(6 s时间内完成充电和放电),电极仍具有44 mAh·g-1比容量。Duan等70对比分析了非纳米NVP颗粒,碳包覆非纳米NVP颗粒(NVP@C)和碳包覆纳米NVP颗粒(nano NVP@C)进行电化学反应时的模型,如图22。离子扩散在非纳米NVP颗粒中距离有限,钠离子只能在颗粒表面进行脱嵌,同时电子传输也由于较差的电导性而受阻。NVP@C虽然在一定程度上提高了电子导电能力,但颗粒内部仍具有未进行电化学反应的部分。相比之下,纳米NVP颗粒被碳包覆形成核壳结构时,碳层能够提供离子和电子快速传输网络,并且纳米NVP颗粒可以因其较小的尺寸而能充分参与电化学反应,产生更好的储能特性。Li等79将NVP纳米颗粒分别与活性炭(AC),一维碳纳米管(CVT),石墨进行了复合并对比了在钠离子半电池体系中的储能特性。NVP/ AC复合物表现出了最优的储钠特性,在0.5C下具有117.5 mAh·g-1比容量,并且5C下循环200次容量保持率为96.4%。NVP/AC电极与AC电极构成的非对称钠离子电池,1C下充放电循环200次后容量保持率为80%。Wang等80采用水热和溶胶凝胶法制备了NVP与多壁碳纳米管(MWCNT)复合物NVP/ MWCNT。在0.2C电流密度下,NVP/MWCNT作为锂离子电池正极产生了82.2 mAh·g-1比容量,100个循环后保持有72.3 mAh·g-1。Shen等81近期报道了氮掺杂碳包覆NVP并与MWCNT复合作为钠离子电池正极的研究,该结构中NVP颗粒被双层碳材料包覆,其倍率和循环性能都得到了较大提升。在0.2C倍率下,该材料初始容量为94.5 mAh· g-1,70C下具有70 mAh·g-1,30C下充放电循环300次后容量保持率为87%。
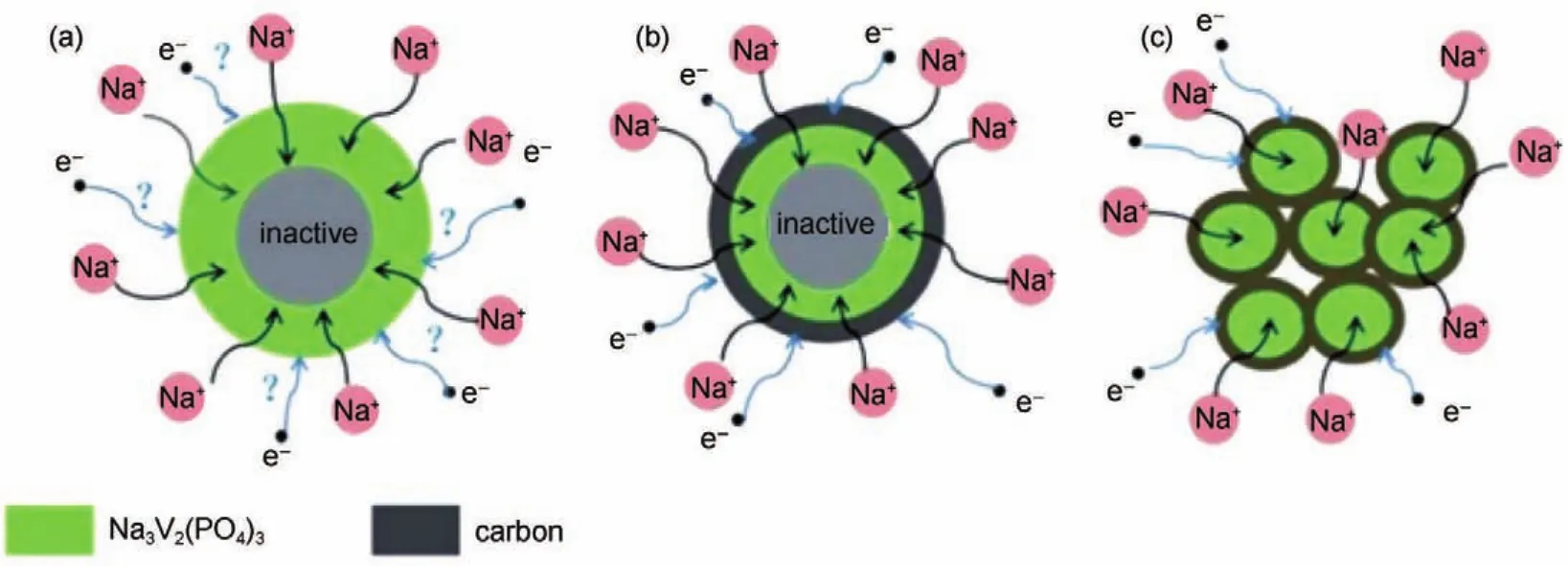
图22 (a)NVP非纳米颗粒,(b)碳包覆的NVP非纳米颗粒和(c)碳包覆的纳米NVP颗粒进行钠离子和电子传导模型70Fig.22 Electron and sodium ion conduction in(a)NVPparticles,(b)NVP@C and(c)nano NVP@C70

图23 (a)三维层级多孔NVP/C/rGO正极有利于钠离子和电子传输;(b)冷冻干燥法合成NVP/C/rGO示意图83Fig.23 (a)3D hierarchical NVP/C/rGO with meso-and macro-pore provides pathways for Na ions and electrons; (b)schematic of freeze-drying-assisted method to prepare NVP/C/rGO83
4.1.2 石墨化碳包覆
颗粒表面的碳包覆一般通过热裂解碳骨架的有机结构形成,并且包覆碳为无定型结构,其导电性能有限。为了进一步提高包覆碳的导电性,Fang等82通过化学气相沉积(CVD)方法将经过高温煅烧得到的NVP颗粒置于CVD管式炉中,原位包覆了石墨烯结构的碳(或称为石墨化的碳)并被同时形成的石墨纤维连接在一起。这种多层级碳包覆的NVP作为钠离子电池正极材料,在500C(58.5 A·g-1)电流密度下仍具有38 mAh·g-1比容量;在30C下经过20000个循环,容量保持率为54%。
4.1.3 石墨烯复合
石墨烯作为一类具有特殊结构的碳材料,已被广泛用作电极材料以及作为基体进行材料负载以提升电导性62,84-88。Yan等83在2015年报道了结合冷冻干燥法,将碳包覆的NVP颗粒包裹在还原的石墨烯(rGO)中形成具有三维层级结构的NVP/C/ rGO复合物(图23a)。图23(b)为NVP/C/rGO的合成过程。聚乙烯吡咯烷酮(PVP)不仅在原料的混合过程中作为螯合剂,使无机盐原料与氧化石墨烯均匀融合,而且在后续的高温煅烧过程中形成多孔碳承载NVP纳米颗粒,有效阻止NVP颗粒在高温反应过程中团聚和生长。纳米NVP颗粒表面包覆有无定型介孔碳,可缩短钠离子固相扩散距离。大孔和介孔结构能够为大电流密度下快速钠离子和电子传导提供网络,并能缓解钠离子脱嵌反应产生的形变。NVP/C/rGO用作钠离子电池正极在1C-20C电流密度下具有115 mAh·g-1的放电比容量,100C下仍保持有86 mAh·g-1。100C下经过10000个循环,容量保持率为64%。Jung等89采用溶胶凝胶与固态烧结方法制备了NVP/石墨烯复合材料,在5C倍率下具有86.3 mAh·g-1比容量,充放电极化电压仅为0.15 V。Guo等90以Na2EDTA作为钠源和氮化碳前驱体在NVP颗粒表面制得原位包覆的氮化碳,与还原氧化石墨烯(rGO)复合后作为钠离子电池正极,0.1C和30C下比容量分别116.8和80 mAh·g-1。Xu等91通过修饰NVP溶胶前驱体电荷,制备了层层组装的NVP纳米晶/rGO复合结构。该复合物中含有3%(w)的rGO和0.5%(w)的无定型碳,在钠离子电池中100C下具有73 mAh·g-1比容量,50C下循环15000次容量保持率为70%。
Zhang等92将NVP纳米晶均匀分散在rGO纳米片中并制成纸状柔性电极。该电极无额外导电剂、粘结剂、集流体,可以直接用于组装电池。该柔性电极在100 mA·g-1电流密度下具有113 mAh·g-1比容量,120个循环后容量保持率为96.6%。他们继续制备了Sb/rGO纸状柔性电极,并与NVP/rGO柔性电极组装为全电池体系,100 mA·g-1电流密度下经过100个充放电循环,该全电池仍保持有400 mAh·g-1比容量。Choi等93采用溶胶凝胶法制备了NVP/Ag+/GS复合电极,Ag+与GS的协同作用提高了NVP颗粒之间的导电性,使其在0.1C下具有102 mAh·g-1比容量,并且5C下可以保持有73 mAh·g-1。
4.1.4 碳基体负载
Jiang等94将NVP纳米颗粒固定在三维高定向介孔碳CMK-3中,CMK-3不仅能够在高温反应阶段阻止NVP颗粒的生长,使其具有纳米尺寸以减小离子扩散路径;还能够作为导电基体,提供快速电子传输网络。在合成过程中,葡萄糖与原料前驱体混合以实现NVP表面碳包覆,形成NVP@C结构。在CMK-3中煅烧后,得到二维方向的“核壳”结构NVP@C@CMK-3。NVP@C@ CMK-3作为钠离子电池正极,在30C电流下比容量可达到81 mAh·g-1;5C电流密度充放,经过2000个循环容量具有78 mAh·g-1。Chu等95采用水热、溶胶凝胶、高温煅烧方法将碳包覆的NVP颗粒,并将其与多壁碳纳米管(MWCNT)进行负载,得到NVP@C/MWCNT复合物。相互交联的MWCNT能够提高NVP颗粒之间的导电性。在钠离子电池体系中,10C倍率下具有81.7 mAh·g-1比容量,循环500次后容量保持率为93.9%。

图24 钾离子掺入NVP6b钠位结构示意图96Fig.24 Representation of K-doped NVPby doping into Na(6b)site96
4.2 基于NVP的体相掺杂
杂原子掺杂可以调节NVP组成与结构,从而影响NVP物理化学性质。目前基于NVP的体相掺杂主要为掺入金属元素取代部分Na或V,改变原NVP的体积结构与电导性97。Lim等96将具有更大离子半径的钾离子掺入到NVP结构中,通过延伸NVP的c轴扩大了NVP晶格体积,并产生了更大的钠离子扩散通道,其结构如图24所示。掺入的钾离子能够稳定NVP结构在电化学反应过程中不会产生较大的体积形变,而钾离子自身并不参与反应。通过对比分析,掺入钾离子含量对掺杂NVP电化学储能特性会产生不同影响。含有0.09个K的Na2.91K0.09V2(PO4)3具有较低的电荷转移电阻,较高的钠离子扩散速度以及稳定的结构而成为最优掺杂比例。Na2.91K0.09V2(PO4)3在0.2C下具有110.4 mAh·g-1的比容量,倍率与循环性能相比于未掺杂NVP具有明显的提高。Aragon等98通过溶胶凝胶法和高温烧结制备了铁(Fe3+)掺杂的Na3V2-xFex(PO4)3(0≤x≤0.5)。NVP中的三价态钒被铁部分替代后,由于铁具有更大的离子半径从而使掺杂后的结构晶胞体积增大。的电化学循环伏安测试中,NVP在0.5 mV·s-1下具有3.5 V(vs Na/Na+)的氧化峰和3.2 V(vs Na/Na+)还原峰,掺杂后的物相在4 V(vs Na/Na+)左右额外出现了一对氧化还原峰,并且随着掺杂量的增加,氧化峰出现正移还原峰负移。放电电位的减小归结于Fe3+取代V3+后所出现的诱导效应,并且由于存在价带的重合,放电过程中没有单独表现出铁的价态变化。X射线光电子能谱表明了在4 V处的电位对应于Na3V2-xFex(PO4)3中V5+/V4+转化,伴随该反应脱嵌的钠离子主要发生在Na(1)位。掺杂后的Na3V2-xFex(PO4)3具有更高的比容量(115 mAh·g-1)和循环稳定性。
Lalere等99通过铝(Al)掺杂NVP,得到了Na3AlyV2-y(PO4)3(y=0.1,0.25,0.5)不同掺杂比的物相。他们发现掺杂后的体系与未掺杂的NVP从低温(-50°C)到高温(250°C)过渡的过程中,其晶体均会发生从单斜系C2到三方系R3¯c的转换。通过对掺杂相晶胞参数进行研究,发现在低温和高温下的物相晶胞参数均随V掺杂比的增加而减小,晶胞体积发生收缩。在1.3-1.8 V之间的钠离子脱嵌反应,主要对应V3+/V2+氧化还原反应,并且随着掺杂比的增加,Na3AlyV2-y(PO4)3氧化还原峰负移,即需要更低的放电电压使钠离子嵌入高掺杂比的物相中。V被Al部分取代后,导致V―O键键长减小,共价键作用增大致使V3+/V2+氧化还原反应变难。

图25 恒电流0.05C下Na3AlyV2-y(PO4)3电极在不同电压区间进行钠离子脱嵌对应的充放电曲线99Fig.25 Electrochemical signatures of Na3AlyV2-y(PO4)3upon the insertion and extraction of sodium ions in specific voltage range at 0.05C99
图25左侧为高电位区间内的钠离子脱嵌反应,主要对应V4+/V3+氧化还原反应。由于部分V被Al取代而不能参与氧化还原反应,致使脱嵌钠离子量降低,比容量也从117.6 mAh·g-1(NVP)降至90.6 mAh·g-1[Na3Al0.5V1.5(PO4)3]。掺杂相中V4+/V3+氧化还原反应峰被分为间距为0.04 V的两对峰,说明发生的两步氧化还原反应。现场原位XRD验证了钠离子迁出Na3Al0.25V1.75(PO4)3到Na1.25Al0.25V1.75(PO4)3的过程中,会出现V4+/V3+混合态的独立相Na2.1Al0.25V1.75(PO4)3。V5+/V4+氧化还原反应发生在3.95 V的平台。Na3Al0.5V1.5(PO4)3充电过程中在3.37V处V3+氧化为V4+,得到Na1.5Al0.5V1.5(PO4)3。继续氧化时其中0.5个V被氧化为V5+,1个V仍为V4+,得到NaAl0.5V1.5(PO4)3。因此Al取代部分V后,可以使NVP中部分V3+分步氧化为V5+,提高工作电压,储能密度从396.3 Wh·kg-1(NVP)提升到424.6 Wh· kg-1[Na3Al0.5V1.5(PO4)3]。Aragon等100报道了铬(Cr)掺杂的Na3CrxV2-x(PO4)3并将其用于钠离子电池。Cr掺杂到NVP结构,减小了NVP中原晶胞参数而不参与电化学反应。掺杂Cr后的体系在4V左右出现了高电压平台,对应体系中V5+/V4+氧化还原反应。Na3Cr0.1V1.9(PO4)3充放电过程电极极化小,0.5C下经过40次充放电后容量保持为107 mAh·g-1,库伦效率为99%。

图26 钠离子脱嵌反应发生在(a)块体材料和(b)一维定向生长颗粒时产生的不同变化73Fig.26 Schematic representation of sodium insertion/deinsertion for(a)bulk and(b)oriented nanoparticles confined in one dimension73
4.3 NVP形貌控制
Kajiyama等73采用静电纺丝技术,制备了碳包覆的NVP一维纳米纤维。静电纺丝是制备一维结构材料的方法,他们将制取NVP的原料混合螯合剂与高分子聚合物后得到前驱体浆料,经静电纺丝产生包含前驱原料的纳米纤维,经过高温煅烧得到碳包覆NVP纤维。该结构中无定型碳厚度约为50 nm,纤维中心为团聚的NVP纳米晶,直径约为20-50 nm。选区电子衍射说明NVP纳米晶在一维碳鞘内呈现一维定向生长特点。纳米颗粒或静电纺丝中由于NVP在形成的过程中经历较多的晶体生长过程,因此很难在一维碳模板内快速定向生长,因而需要较长的高温反应时间以实现高定向NVP晶体的生长。该一维结构中碳鞘能与NVP中的聚阴离子体结构形成C―O键等稳定的化学键,并可以有效阻止内部NVP副反应的发生。如图26所示,块体材料在钠离子脱嵌时会产生较大的体积形变,内部还会出现裂缝缺陷结构。定向生长的纳米颗粒可以缓解钠离子脱嵌时产生的应力,并有效缩短了离子和电子传导路径,利于提高材料的循环寿命和倍率特性。Li等101采用静电纺丝法得到纤维前驱体后经过二次煅烧,得到一维NVP/C纳米棒结构。NVP/C纳米棒用作钠离子电池正极在0.05C下产生了116.9 mAh·g-1比容量,0.5C下具有105.3 mAh·g-1比容量。Liu等102通过制备静电纺丝技术,将NVP纳米颗粒均匀包裹在一维的交联碳纤维中,得到NVP/C的复合电极。该电极在0.1C倍率下具有101 mAh·g-1放电容量,并且由于材料的高电导性网络,高倍率电流下仍能保持理想比容量。
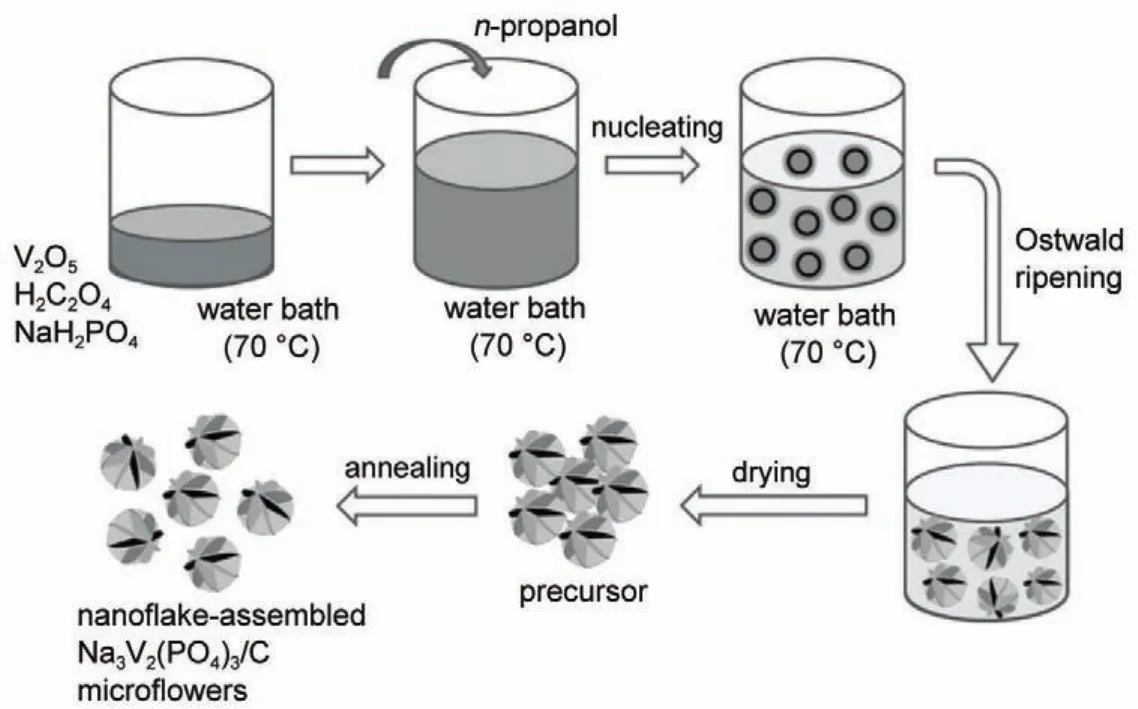
图27 多层级NVP/C微米花合成示意图50Fig.27 Schematic routes for preparation of nanoflake-assembled NVP/C hierarchical microflowers50
An等50通过共沉淀方法制备了纳米片组装的多层级NVP/C微米花状结构,示意图如图27所示。多层级NVP/C微米花结构除了使其导电性得到提升以外,还增大了电极与电解液的接触面积,并缩短了离子扩散距离。750°C煅烧后的材料与锂负极和锂有机电解液构成混合离子电池体系具有最优的电化学性能,An等分别研究了不同电压区间内的储能特性:在电流密度为0.91C,电压区间1-4.3 V内产生了230 mAh·g-1比容量,45C下达130 mAh·g-1,200个充放电循环后容量保持为138 mAh·g-1;在电压区间2.5-4.3 V内,0.91C下首次容量为103 mAh·g-1,91C下容量为43 mAh·g-1,9.1C下充放电循环5000次,容量保持率为83.6%;在电压区间1-2.5 V内,0.91C下首次容量为98 mAh·g-1,91C下容量为72 mAh·g-1,9.1C下充放电循环7000次,容量保持率为97.5%。
5 总结与展望
NVP作为一类钠离子超导体,具有很高的钠离子扩散速率。基元结构可在3.4 V(vs Na/Na+)电压下发生两个钠离子的脱嵌,产生117 mAh·g-1的比容量;通过1.6 V vs(Na/Na+)和0.3 V vs(Na/Na+)两处电压平台,两个钠离子嵌入体系形成Na4V2(PO4)3和Na5V2(PO4)3,分别产生约59和54 mAh·g-1的比容量。因此,NVP是一个可以同时提供高压脱钠、低压嵌钠并具有较大克容量的材料,为此可用作对称电池电极。通过金属原子掺杂,能提高脱嵌离子电位,从而有助于提升材料的储能密度,同时也有助于减小钠离子脱嵌过程中的体积形变。NVP稳定的“灯笼式”结构使其具有很高的热稳定性,能够适用于高温电池设计,但该结构同时也导致了较低的电导率。针对NVP的研究也主要集中在如何显著提高其电导率,以提升在大倍率电流充放和长循环过程中的稳定性。
关于NVP的大多数研究为室温下储能特性,尽管有相关研究报道了其低温下会转换为不同晶型,但低温下其电化学反应机理和储钠性能尚未研究透彻。低温NVP具有单斜对称特点,钠离子呈现有序排布,因此有望用于设计低温钠离子电池;由于电解液分解电压的限制,NVP的工作电压一般低于4.6 V(vs Na/Na+),无法用实验观测其Na(1)位占据离子的变化。关于Na(1)位钠离子能否发生脱嵌仍存在较多争议,尽管有V2(PO4)3的相关报道,但很多研究表明在有限的电压区间内,仅能实现Na(2)位离子的脱嵌;NVP在低压区间(<2 V)可以允许额外钠离子嵌入,形成NaxV2(PO4)3(x>3),并在低电位区间进行可以脱嵌,但相关实验和机理研究较少;作为一类嵌入型材料,NVP在充放电过程中仍会产生较大的体积形变而影响循环性能。同时,一种低成本、易操作并能显著提升电导率的方法仍需完善。NVP的结构特点使其可用作为正、负极,适用于高温、室温、低温环境,NVP在不同工作状态下的储能机理研究对于进一步认识NVP结构特性,提升性能具有重要意义。
(1)Larcher,D.;Tarascon,J.M.Nat.Chem.2015,7,19. doi:10.1038/nchem.2085
(2)Tarascon,J.M.;Armand,M.Nature 2001,414,359. doi:10.1038/35104644
(3)Armand,M.;Tarascon,J.M.Nature 2008,451,652. doi:10.1038/451652a
(4)Dunn,B.;Kamath,H.;Tarascon,J.M.Science 2011,334,928. doi:10.1126/science.1212741
(5)Yang,Z.;Zhang,J.;Kintner-Meyer,M.C.W.;Lu,X.;Choi, D.;Lemmon,J.P.;Liu,J.Chem.Rev.2011,111,3577. doi:10.1021/cr100290v
(6)Saravanan,K.;Mason,C.W.;Rudola,A.;Wong,K.H.; Balaya,P.Adv.Energy Mater.2013,3,444.doi:10.1002/ aenm.201200803
(7)Palomares,V.;Serras,P.;Villaluenga,I.;Hueso,K.B.; Carretero-González,J.;Rojo,T.Energ.Environ.Sci.2012,5, 5884.doi:10.1039/c2ee02781j
(8)Ong,S.P.;Chevrier,V.L.;Hautier,G.;Jain,A.;Moore,C.; Kim,S.;Ma,X.;Ceder,G.Energ.Environ.Sci.2011,4,3680. doi:10.1039/c1ee01782a
(9)Raju,V.;Rains,J.;Gates,C.;Luo,W.;Wang,X.;Stickle,W. F.;Stucky,G.D.;Ji,X.Nano Lett.2014,14,4119.doi: 10.1021/nl501692p
(10)Slater,M.D.;Kim,D.;Lee,E.;Johnson,C.S.Adv.Funct. Mater.2013,23,947.doi:10.1002/adfm.201200691
(11)Li,Z.;Young,D.;Xiang,K.;Carter,W.C.;Chiang,Y.-M.Adv. Energy Mater.2013,3,290.doi:10.1002/aenm.201200598
(12)Palomares,V.;Casas-Cabanas,M.;Castillo-Martinez,E.;Han, M.H.;Rojo,T.Energ.Environ.Sci.2013,6,2312. doi:10.1039/c3ee41031e
(13)Pan,H.;Hu,Y.S.;Chen,L.Energ.Environ.Sci.2013,6, 2338.doi:10.1039/c3ee40847g
(14)Yabuuchi,N.;Kubota,K.;Dahbi,M.;Komaba,S.Chem.Rev.2014,114,11636.doi:10.1021/cr500192f
(15)Wang,L.;Lu,Y.;Liu,J.;Xu,M.;Cheng,J.;Zhang,D.; Goodenough,J.B.Angew.Chem.Int.Ed.2013,52,1964. doi:10.1002/anie.201206854
(16)Kundu,D.;Talaie,E.;Duffort,V.;Nazar,L.F.Angew.Chem. Int.Ed.Engl.2015,54,3431.doi:10.1002/anie.201410376
(17)Kim,S.W.;Seo,D.H.;Ma,X.;Ceder,G.;Kang,K.Adv. Energy Mater.2012,2,710.doi:10.1002/aenm.201200026
(18)Kim,H.;Hong,J.;Park,K.Y.;Kim,H.;Kim,S.W.;Kang,K. Chem.Rev.2014,114,11788.doi:10.1021/cr500232y
(19)Chen,J.;Hou,H.;Yang,Y.;Song,W.;Zhang,Y.;Yang,X.; Lan,Q.;Ji,X.Electrochim.Acta 2015,164,330.doi:10.1016/ j.electacta.2015.02.202
(20)Ye,F.P.;Wang,L.;Lian,F.;He,X.M.;Tian,G.Y.;Ouyang, M.G.Chem.Eng.Prog.2013,1789.[叶飞鹏,王 莉,连芳,何向明,田光宇,欧阳明高.化工进展,2013,1789.]doi: 10.3969/j.issn.1000-6613.2013.08.012
(21)Zhang,N.;Liu,Y.C.;Chen,C.C.;Tao,Z.L.;Chen,J.Chin.J. Inorg.Chem.2015,31,1739.[张 宁,刘永畅,陈程成,陶占良,陈 军.无机化学学报,2015,31,1739.]doi:10.11862/ cjic.2015.258
(22)Cao,Y.;Luo,X.;Yu,H.;Peng,F.;Wang,H.;Ning,G. Catalysis Science&Technology 2013,3,2654.doi:10.1039/ c3cy00256j
(23)Komaba,S.;Murata,W.;Ishikawa,T.;Yabuuchi,N.;Ozeki,T.; Nakayama,T.;Ogata,A.;Gotoh,K.;Fujiwara,K.Adv.Funct. Mater.2011,21,3859.doi:10.1002/adfm.201100854
(24)Park,Y.U.;Seo,D.H.;Kwon,H.S.;Kim,B.;Kim,J.;Kim, H.;Kim,I.;Yoo,H.I.;Kang,K.J.Am.Chem.Soc.2013,135, 13870.doi:10.1021/ja406016j
(25)Goodenough,J.B.;Hong,H.Y.P.;Kafalas,J.A.Materials Research Bulletin 1976,11,203.doi:10.1016/0025-5408(76) 90077-5
(26)Tripathi,R.;Wood,S.M.;Islam,M.S.;Nazar,L.F.Energ. Environ.Sci.2013,6,2257.doi:10.1039/c3ee40914g
(27)Li,J.;Daniel,C.;Wood,D.J.Power.Sources 2011,196,2452. doi:10.1016/j.jpowsour.2010.11.001
(28)Kabbour,H.;Coillot,D.;Colmont,M.;Masquelier,C.; Mentre,O.J.Am.Chem.Soc.2011,133,11900.doi:10.1021/ ja204321y
(29)Ferrari,A.C.;Robertson,J.Phys.Rev.B 2001,63,121405. doi:10.1103/PhysRevB.63.121405
(30)Park,S.I.;Gocheva,I.;Okada,S.;Yamaki,J.I.J. Electrochem.Soc.2011,158,A1067.doi:10.1149/1.3611434
(31)Wang,D.;Liu,Q.;Chen,C.;Li,M.;Meng,X.;Bie,X.;Wei, Y.;Huang,Y.;Du,F.;Wang,C.;Chen,G.ACS Appl.Mater. Interfaces 2016,8,2238.doi:10.1021/acsami.5b11003
(32)Shakoor,R.A.;Seo,D.H.;Kim,H.;Park,Y.U.;Kim,J.;Kim, S.W.;Gwon,H.;Lee,S.;Kang,K.J.Mater.Chem.2012,22, 20535.doi:10.1039/C2JM33862A
(33)Song,W.;Liu,S.Solid State Sciences 2013,15,1. doi:10.1016/j.solidstatesciences.2012.09.012
(34)Song,W.;Ji,X.;Wu,Z.;Zhu,Y.;Li,F.;Yao,Y.;Banks,C.E. RSC Adv.2014,4,11375.doi:10.1039/c3ra47878e
(35)Jiang,T.;Chen,G.;Li,A.;Wang,C.Z.;Wei,Y.J.J.Alloy. Compd.2009,478,604.
(36)Song,W.;Ji,X.;Pan,C.;Zhu,Y.;Chen,Q.;Banks,C.E.Phys. Chem.Chem.Phys.2013,15,14357.doi:10.1039/c3cp52308j
(37)Jian,Z.;Han,W.;Lu,X.;Yang,H.;Hu,Y.S.;Zhou,J.;Zhou, Z.;Li,J.;Chen,W.;Chen,D.;Chen,L.Adv.Energy Mater. 2013,3,156.doi:10.1002/aenm.201200558
(38)Jian,Z.;Zhao,L.;Pan,H.;Hu,Y.S.;Li,H.;Chen,W.;Chen, L.Electrochem.Commun.2012,14,86.doi:10.1016/j. elecom.2011.11.009
(39)Gopalakrishnan,J.;Rangan,K.K.Chem.Mater.1992,4,745. doi:10.1021/cm00022a001
(40)Delmas,C.;Olazcuaga,R.;Cherkaoui,F.;Brochu,R.;Le Flem,G.C.R.Seances Acad.Sci.,Ser.C 1978,287,169.
(41)Cushing,B.L.;Goodenough,J.B.J.Solid State Chem.2001, 162,176.doi:10.1006/jssc.2001.9213
(42)Zatovsky,I.V.Acta Crystallographica.Section E,Structure Reports Online 2010,66,i12.
(43)Lim,S.Y.;Kim,H.;Shakoor,R.A.;Jung,Y.;Choi,J.W.J. Electrochem.Soc.2012,159,A1393.doi:10.1149/2.015209jes
(44)Jian,Z.;Yuan,C.;Han,W.;Lu,X.;Gu,L.;Xi,X.;Hu,Y.S.; Li,H.;Chen,W.;Chen,D.;Ikuhara,Y.;Chen,L.Adv.Funct. Mater.2014,24,4265.doi:10.1002/adfm.201400173
(45)Chotard,J.N.;Rousse,G.;David,R.;Mentré,O.;Courty,M.; Masquelier,C.Chem.Mater.2015,27,5982.doi:10.1021/acs. chemmater.5b02092
(46)Lalère,F.;Leriche,J.B.;Courty,M.;Boulineau,S.;Viallet,V.; Masquelier,C.;Seznec,V.J.Power.Sources 2014,247,975. doi:10.1016/j.jpowsour.2013.09.051
(47)Gaubicher,J.;Wurm,C.;Goward,G.;Masquelier,C.;Nazar, L.Chem.Mater.2000,12,3240.doi:10.1021/cm000345g
(48)Chen,C.;Wen,Y.;Hu,X.;Ji,X.;Yan,M.;Mai,L.;Hu,P.; Shan,B.;Huang,Y.Nat.Commun.2015,6.doi:10.1038/ ncomms7929
(49)Pivko,M.;Arcon,I.;Bele,M.;Dominko,R.;Gaberscek,M. J.Power.Sources 2012,216,145.doi:10.1016/j. jpowsour.2012.05.037
(50)An,Q.;Xiong,F.;Wei,Q.;Sheng,J.;He,L.;Ma,D.;Yao,Y.; Mai,L.Adv.Energy Mater.2015,5.doi:10.1002/ aenm.201401963
(51)Du,K.;Guo,H.;Hu,G.;Peng,Z.;Cao,Y.J.Power.Sources 2013,223,284.doi:10.1016/j.jpowsour.2012.09.069
(52)Song,W.;Cao,X.;Wu,Z.;Chen,J.;Huangfu,K.;Wang,X.; Huang,Y.;Ji,X.Phys.Chem.Chem.Phys.2014,16,17681. doi:10.1039/c4cp01821d
(53)Cong,H.P.;He,J.J.;Lu,Y.;Yu,S.H.Small 2010,6,169. doi:10.1002/smll.200901360
(54)Song,W.;Ji,X.;Wu,Z.;Zhu,Y.;Yang,Y.;Chen,J.;Jing,M.;Li,F.;Banks,C.E.J.Mater.Chem.A 2014,2,5358. doi:10.1039/c4ta00230j
(55)Kang,J.;Baek,S.;Mathew,V.;Gim,J.;Song,J.;Park,H.; Chae,E.;Rai,A.K.;Kim,J.J.Mater.Chem.2012,22,20857.
(56)Anantharamulu,N.;Koteswara Rao,K.;Rambabu,G.;Vijaya Kumar,B.;Radha,V.;Vithal,M.J.Mater.Sci.2011,46,2821. doi:10.1007/s10853-011-5302-5
(57)Yaroslavtsev,A.B.;Stenina,I.A.Russian Journal of Inorganic Chemistry 2006,51,S97.doi:10.1134/s0036023606130043
(58)Song,H.-K.;Lee,K.T.;Kim,M.G.;Nazar,L.F.;Cho,J.Adv. Funct.Mater.2010,20,3818.doi:10.1002/adfm.201000231
(59)Song,W.;Ji,X.;Wu,Z.;Zhu,Y.;Yao,Y.;Huangfu,K.;Chen, Q.;Banks,C.E.J.Mater.Chem.A 2014,2,2571.doi:10.1039/ c3ta14472k
(60)Rui,X.H.;Ding,N.;Liu,J.;Li,C.;Chen,C.H.Electrochim. Acta 2010,55,2384.doi:10.1016/j.electacta.2009.11.096
(61)Yang,Y.;Qiao,B.;Yang,X.;Fang,L.;Pan,C.;Song,W.;Hou, H.;Ji,X.Adv.Funct.Mater.2014,24,4349.doi:10.1002/ adfm.201304263
(62)Song,W.;Ji,X.;Yao,Y.;Zhu,H.;Chen,Q.;Sun,Q.;Banks, C.E.Phys.Chem.Chem.Phys.2014,16,3055.doi:10.1039/ c3cp54604g
(63)Song,W.;Ji,X.;Zhu,Y.;Zhu,H.;Li,F.;Chen,J.;Lu,F.;Yao, Y.;Banks,C.E.ChemElectroChem 2014,1,871.doi:10.1002/ celc.201300248
(64)Santos-Peña,J.;Crosnier,O.;Brousse,T.Electrochim.Acta 2010,55,7511.doi:10.1016/j.electacta.2009.12.069
(65)Lee,J.W.;Hong,J.K.;Kjeang,E.Electrochim.Acta 2012,83, 430.doi:10.1016/j.electacta.2012.07.104
(66)Reddy,R.N.;Reddy,R.G.J.Power.Sources 2003,124,330. doi:10.1016/s0378-7753(03)00600-1
(67)Reddy,R.N.;Reddy,R.G.J.Power.Sources 2006,156,700. doi:10.1016/j.jpowsour.2005.05.071
(68)Wang,S.;Zhao,J.;Wang,L.;Liu,X.;Wu,Y.;Xu,J.Ionics 2015,21,2633.doi:10.1007/s11581-015-1428-9
(69)Noguchi,Y.;Kobayashi,E.;Plashnitsa,L.S.;Okada,S.; Yamaki,J.I.Electrochim.Acta 2013,101,59.doi:10.1016/j. electacta.2012.11.038
(70)Duan,W.;Zhu,Z.;Li,H.;Hu,Z.;Zhang,K.;Cheng,F.;Chen, J.J.Mater.Chem.A 2014,2,8668.doi:10.1039/c4ta00106k
(71)Plashnitsa,L.S.;Kobayashi,E.;Noguchi,Y.;Okada,S.; Yamaki,J.I.J.Electrochem.Soc.2010,157,A536.doi: 10.1149/1.3298903
(72)Jian,Z.;Sun,Y.;Ji,X.Chem.Commun.2015,51,6381.doi: 10.1039/C5CC00944H
(73)Kajiyama,S.;Kikkawa,J.;Hoshino,J.;Okubo,M.;Hosono, E.Chem.-Eur.J.2014,20,12636.doi:10.1002/ chem.201403126
(74)Song,W.;Wu,Z.;Chen,J.;Lan,Q.;Zhu,Y.;Yang,Y.;Pan,C.; Hou,H.;Jing,M.;Ji,X.Electrochim.Acta 2014,146,142. doi:10.1016/j.electacta.2014.09.068
(75)Song,W.;Cao,X.;Wu,Z.;Chen,J.;Zhu,Y.;Hou,H.;Lan,Q.; Ji,X.Langmuir 2014,30,12438.doi:10.1021/la5025444
(76)Song,W.;Ji,X.;Wu,Z.;Yang,Y.;Zhou,Z.;Li,F.;Chen,Q.; Banks,C.E.J.Power.Sources 2014,256,258.doi:10.1016/j. jpowsour.2014.01.025
(77)Chen,Z.;Dai,C.;Wu,G.;Nelson,M.;Hu,X.;Zhang,R.;Liu, J.;Xia,J.Electrochim.Acta 2010,55,8595.doi:10.1016/j. electacta.2010.07.068
(78)Zhu,C.;Song,K.;VanAken,P.A.;Maier,J.;Yu,Y.Nano Lett. 2014,14,2175.doi:10.1021/nl500548a
(79)Li,S.;Dong,Y.;Xu,L.;Xu,X.;He,L.;Mai,L.Adv.Mater. 2014,26,3545.doi:10.1002/adma.201305522
(80)Wang,W.J.;Zhao,H.B.;Yuan,A.B.;Fang,J.H.;Xu,J.Q. Acta Phys.-Chim.Sin.2014,30,1113.[王文俊,赵宏滨,袁安保,方建慧,徐甲强.物理化学学报,2014,30,1113.] doi:10.3866/PKU.WHXB201404182.
(81)Shen,W.;Li,H.;Guo,Z.;Wang,C.;Li,Z.;Xu,Q.;Liu,H.; Wang,Y.;Xia,Y.ACS Appl.Mater.Interfaces.2016,8,15341. doi:10.1021/acsami.6b03410
(82)Fang,Y.;Xiao,L.;Ai,X.;Cao,Y.;Yang,H.Adv.Mater.2015, 27,5895.doi:10.1002/adma.201502018
(83)Rui,X.;Sun,W.;Wu,C.;Yu,Y.;Yan,Q.Adv.Mater.2015,27, 6670.doi:10.1002/adma.201502864
(84)Chen,D.;Tang,L.;Li,J.Chem.Soc.Rev.2010,39,3157.doi: 10.1039/B923596E
(85)Huang,X.;Qi,X.;Boey,F.;Zhang,H.Chem.Soc.Rev.2012, 41,666.doi:10.1039/c1cs15078b
(86)Song,W.;Ji,X.;Deng,W.;Chen,Q.;Shen,C.;Banks,C.E. Phys.Chem.Chem.Phys.2013,15,4799.doi:10.1039/ C3CP50516B
(87)Song,W.;Chen,J.;Ji,X.;Zhang,X.;Xie,F.;Riley,D.J.J. Mater.Chem.A 2016,4,8762.doi:10.1039/C6TA02548J
(88)Yang,Y.;Ji,X.;Yang,X.;Wang,C.;Song,W.;Chen,Q.; Banks,C.E.RSC Adv.2013,3,16130.doi:10.1039/ c3ra43010c
(89)Jung,Y.H.;Lim,C.H.;Kim,D.K.J.Mater.Chem.A 2013,1, 11350.doi:10.1039/c3ta12116j
(90)Guo,J.Z.;Wu,X.L.;Wan,F.;Wang,J.;Zhang,X.H.;Wang, R.S.Chem.-Eur.J.2015,21,17371.doi:10.1002/ chem.201502583
(91)Xu,Y.;Wei,Q.;Xu,C.;Li,Q.;An,Q.;Zhang,P.;Sheng,J.; Zhou,L.;Mai,L.Adv.Energy Mater.2016,doi:10.1002/ aenm.201600389
(92)Zhang,W.;Liu,Y.;Chen,C.;Li,Z.;Huang,Y.;Hu,X.Small 2015,11,3822.doi:10.1002/smll.201500783
(93)Choi,M.S.;Kim,H.S.;Lee,Y.M.;Lee,S.M.;Jin,B.S.J Nanosci.Nanotechno.2015,15,8937.doi:10.1166/ jnn.2015.11538
(94)Jiang,Y.;Yang,Z.;Li,W.;Zeng,L.;Pan,F.;Wang,M.;Wei, X.;Hu,G.;Gu,L.;Yu,Y.Adv.Energy Mater.2015,5, 1402104.doi:10.1002/aenm.201402104
(95)Chu,Z.;Yue,C.Solid State Ionics 2016,287,36.doi:10.1016/ j.ssi.2015.07.024
(96)Lim,S.J.;Han,D.W.;Nam,D.H.;Hong,K.S.;Eom,J.Y.; Ryu,W.H.;Kwon,H.S.J.Mater.Chem.A 2014,2,19623. doi:10.1039/C4TA03948C
(97)Mouahid,F.E.;Zahir,M.;Maldonado-Manso,P.;Bruque,S.; Losilla,E.R.;Aranda,M.A.G.;Rivera,A.;Leon,C.; Santamaria,J.J.Mater.Chem.2001,11,3258.doi:10.1039/ B102918P
(98)Aragón,M.J.;Lavela,P.;Ortiz,G.F.;Tirado,J.L. J.Electrochem.Soc.2015,162,A3077.doi:10.1149/ 2.0151502jes
(99)Lalere,F.;Seznec,V.;Courty,M.;David,R.;Chotard,J.N.; Masquelier,C.J.Mater.Chem.A 2015,3,16198.doi:10.1039/ C5TA03528G
(100)Aragón,M.J.;Lavela,P.;Ortiz,G.F.;Tirado,J.L. ChemElectroChem 2015,2,995.doi:10.1002/celc.201500052
(101)Li,H.;Bai,Y.;Wu,F.;Ni,Q.;Wu,C.Solid State Ionics 2015, 278,281.doi:10.1016/j.ssi.2015.06.026
(102)Liu,J.;Tang,K.;Song,K.;vanAken,P.A.;Yu,Y.;Maier,J. Nanoscale 2014,6,5081.doi:10.1039/C3NR05329F
Progress in the Investigation and Application of Na3V2(PO4)3for Electrochemical Energy Storage
SONG Wei-Xin1,2HOU Hong-Shuai1JI Xiao-Bo1,*
(1College of Chemistry and Chemical Engineering,Central South University,Changsha 410083,P.R.China;2Department of Materials,Imperial College London,London SW72AZ,UK)
Lithium ion batteries(LiBs)have been widely utilized,but the limited lithium resource restricts development and application of LiBs in large-scale energy storage.Sodium has similar physicochemical characteristics to that of lithium and is suitable to transfer between two electrodes as a cation in the“rocking chair”mechanism of LiBs.Na-containing compounds have been proposed as the electrodes to store sodium ions and provide channels for diffusion.Polyanion Na3V2(PO4)3is a Na-super-ionic conductor(NASICON)with specific Na sites in its crystal structure and three-dimensional open channels.Recently,Na3V2(PO4)3has been demonstrated as potential electrode material with promising properties for energy storage.In this review we systematically summarize the structure of Na3V2(PO4)3,the application and mechanism in a specific energy system,and the recent development of Na3V2(PO4)3structure for use as electrodes.The potential problems and trends of Na3V2(PO4)3are also discussed.
Na3V2(PO4)3;Na-super-ionic conductor;Electrochemistry;Energy storage;Material structure
O646
iew]
10.3866/PKU.WHXB201608303www.whxb.pku.edu.cn
Received:June 23,2016;Revised:August 26,2016;Published online:August 30,2016.
*Corresponding author.Email:xji@csu.edu.cn;Tel:+86-731-88877237.
The project was supported by the National Natural Science Foundation of China(21473258,21673298,51622406).
国家自然科学基金(21473258,21673298,51622406)资助项目
©Editorial office ofActa Physico-Chimica Sinica
