基于开放染色质的全基因组水平转录调控元件的研究方法与进展
2017-03-07韩金磊李占杰
韩金磊, 李占杰, 王 凯,3
(1.福建农林大学海峡联合研究院基因组与生物技术研究中心,福建 福州 350002;2.福建农林大学作物科学学院,福建 福州 350002;3.福建农林大学国家甘蔗工程技术研究中心,福建 福州 350002)
基于开放染色质的全基因组水平转录调控元件的研究方法与进展
韩金磊1,2, 李占杰1, 王 凯1,2,3
(1.福建农林大学海峡联合研究院基因组与生物技术研究中心,福建 福州 350002;2.福建农林大学作物科学学院,福建 福州 350002;3.福建农林大学国家甘蔗工程技术研究中心,福建 福州 350002)
真核生物转录调控过程是大量的顺式调控元件与反式作用因子相互作用的结果.研究发现,这一调控过程与染色质核小体的动态定位相关,调控因子的结合需要裸露的无核小体的DNA区域,即开放的染色质位点.因此,高效精确地定位基因组上的开放染色质位点为成功地发掘基因组调控元件,乃至揭示基因表达调控机制提供了重要线索和有效手段.本文对开放染色质位点的定义、主要研究方法以及功能注释进行概述,希望对在基因组水平上调控元件的发掘,尤其是在植物中的应用提供借鉴.
开放染色质位点; DNase Ⅰ超敏感位点测序; 调控因子印记; 表观遗传修饰
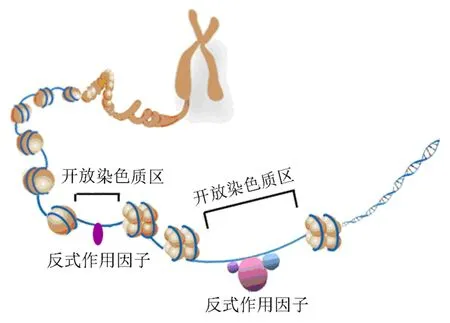
图1 开放染色质模式图Fig.1 Pattern of open chromatin
真核生物染色体是由DNA分子包裹组蛋白形成的核小体结构并通过一系列折叠过程而形成的.作为遗传物质的载体,染色体无时无刻不在进行着转录与复制等生物学过程,而这些过程的顺利进行需要染色体上特定的调控元件与相应的调控蛋白因子正确地结合.研究表明,染色质核小体的定位是动态的[1],而这恰恰与调控因子的结合有直接的联系,即调控因子的结合需要裸露的无核小体结合的DNA部位.基于此,人们将这些裸露的染色质区间称作开放位点(图1),而如何精确定位这些开放的染色质区域也就成为了发掘基因组调控元件,乃至揭示基因表达调控机制的重要线索和有效手段.由于开放染色质较其他部位具有更高的敏感性,更容易发生剪切,因此,人们提出了采用核酸酶、转座酶及物理断裂等方法对染色质进行切割,然后结合不同的序列分析方法,便可以获得这些敏感切割位点的信息.经过长期的摸索,目前在动物上已经建立起了一系列不同的技术方法,如Southern杂交、DNase Ⅰ超敏感位点(DNase I hypersensitive sites, DHs)测序、甲醛辅助的调控元件分离(formaldehyde assisted isolation of regulatory elements, FAIRE)测序、染色质转座酶可接近性(assay for transposase accessible chromatin, ATAC)测序和转座子超敏感位点(transposome hypersensitive sites, THS) 测序,并得到了深入广泛的应用[2],而与之相比,在植物中的相关研究尚处于探索阶段,开展较少.本文将对该技术发展的历程及其应用进行综合阐述,希望为其在动植物,尤其是在植物基因组水平上调控元件的发掘及调控机制的探究等功能基因组研究提供借鉴.
1 开放染色质区的发现及其检测方法
1.1 DHs的定义及研究方法的建立
上世纪70年代末,两个科研团队在研究SV40病毒染色质结构时相继发现该病毒染色质的某些区域对核酸酶表现出不同的切割敏感性.首先,Varshavsky et al[3]采用EcoRⅠ、BamHⅠ和BglⅠ等限制性核酸酶切割SV40的染色体时,发现它们在一定的反应条件下并非随机切割,而是表现出切割位点的偏好性,尤其在复制起始位点最为敏感,并且这一特性与切割经过甲醛固定的染色质相同.不久之后,Scott et al[4]同样发现SV40染色质经过DNase Ⅰ以及一个内源核酸酶的酶切后,其某些区域表现出对内切酶的高度敏感性,而相应的基因组DNA则表现出随机切割的特征.随后,来自美国哈佛大学的另一研究团队采用果蝇染色质为材料,发现DNase Ⅰ和微球菌核酸酶在其染色质上的某些特定区域呈现高度剪切敏感性,从而首次在多细胞真核生物中印证了这一特性[5].为了解释这一现象,他们针对DNase I剪切的结果开展了深入分析.通过Southern杂交方法将DNase Ⅰ剪切后的DNA转移到杂交膜上,并用放射性标记的5个特定基因组DNA做为探针进行检测,结果证实了果蝇染色质某些区域确实存在DNase Ⅰ剪切敏感性;同时,发现这些区域的大小可以从几千碱基对到上万碱基对.更有意义的是,这一研究为基于核酸酶敏感位点的分析研究开创了一个新的方法.在此后很长的一段时间内,人们均是采用Southern杂交的方法开展核酸酶敏感位点的分析工作.
随着相关研究的不断深入,人们发现这些敏感位点在对核酸酶的敏感性上往往较其他区域高出至少1个数量级,核酸酶超敏感位点的定义便由此而来[6].同时,相关研究也主要集中到了DNase Ⅰ这一核酸酶的研究上来[7],并逐渐形成了今天广泛应用的DHs研究策略.更为重要的是,研究发现这些DHs往往与正在进行转录或具有转录潜能的基因相关联[5,8].其中,一个著名的例子就是对果蝇hsp26基因区的DHs的研究[9].热诱导条件下,在果蝇热激蛋白基因hsp26启动子区会出现两个DHs,研究表明,这两个位点即为HSTF与GAGA蛋白的结合位点,它们的结合直接决定着热激条件下hsp26基因的高效转录,同时也印证了DHs与转录因子的直接联系.
到目前为止,众多研究业已证实,DHs通常是顺式调控元件所在的区域,而它对DNase Ⅰ的敏感性主要是由于其与反式作用因子的结合导致的.具体而言,反式作用因子的结合将导致这些区域缺少核小体结构,染色质裸露、结构疏松,易于与DNase Ⅰ结合并发生剪切(图1),从而表现出对DNase Ⅰ的高度敏感性[10].目前已证明,包括启动子、增强子、绝缘子和抑制因子等在内的多种基因调控元件均与DHs紧密关联[11].因此,DHs也被认为是基因调控元件鉴定的“金标准”[12].
早期的DHs主要借助Southern杂交的方法进行分析,但由于需要已知片段作为探针,因此只能针对少数已知的基因组位点进行研究[5,7].2004年,John A. Stamatoyannopoulos和Francis S. Collins两个团队采用类似的DHs克隆的方法,首次在全基因组水平上开展了人类DHs的分析工作[12-13].不久之后,为了提高检测的灵敏度和准确性,Boyle et al借助于第2代测序技术的特点,开创性地将DNase Ⅰ酶解后的DNA片段直接用来进行测序文库的构建,并进行高通量测序,从而快速、准确地从人类CD4+ T细胞中鉴定出9万多个DHs[14](图2).这一新技术的建立使得DHs的鉴定更加简单易行,同时检测的灵敏度和准确度也获得了极大的提升.在此之后,研究者又以这一技术原理为基础发展出了低样品量和单细胞水平的DHs研究新技术[15-16],为精细揭示机体发育机制提供了一个强有力的工具.
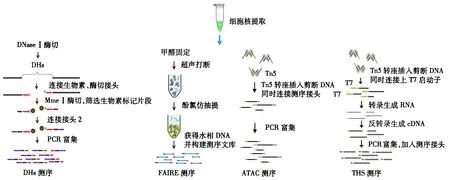
图2 几种高通量检测开放染色质位点的技术方法的流程图Fig.2 Workflows of several high-throughput methods for open-chromatin identification
1.2 基于超声打断的FAIRE测序
FAIRE测序技术是把超声打断与高通量测序技术相结合的开放染色质区鉴定方法[17].它的发现十分偶然,是科研人员在染色质免疫沉淀试验中误操作将制备对照DNA的细胞进行了甲醛交联,进行测序后发现,本应随机分布的序列在编码区表现出富集的特征.进一步分析发现,这些编码区缺少核小体,即开放染色质区,它们易于被打断提取而导致富集.由此,这一方法也被研究者应用到开放染色质区的鉴定上来.该方法经过改进,成熟的步骤主要包括:细胞或组织培养收集、甲醛交联、细胞裂解、超声打断,然后分离出水相DNA,并进行测序及后续分析.其中,交联染色质经超声处理后,采用酚氯仿抽提,打断的DNA由于具有可溶性而被提取出来,从而获得开放染色质的DNA(图2).相对于DHs测序,FAIRE测序操作更简单.但它有一个致命的缺陷,就是该方法中甲醛交联的条件难以衡量确定,交联的程度难以把握.交联过度会导致非特异的信号产生,而交联过轻则会降低检测覆盖度,因此,正确的交联条件需要在不同材料间开展大量的摸索尝试.通过与DHs测序结果比较发现,两种方法鉴定出的开放染色质区重叠度不超过50%[18-19];与转座子Tn5建立的方法相比较,同样存在一致性较低的问题[20].因此,这一缺陷导致了该方法的应用受到了很大的限制.
1.3 基于转座子的ATAC测序
Tn5转座子是复合转座子,由于其转座机制简单,随机性好且插入位点易检测,因此早期被应用于插入突变的研究[21].随着其高频转座突变体的发现,人们看到其在DNA剪切方面的潜力,于是将其应用到高通量测序文库的构建中来[22-23].最初,人们只是用它来剪切DNA并通过其引入测序接头而快速地进行测序文库的构建,但斯坦福大学的Buenrostro等人意识到转座子虽然对于裸露DNA表现出随机插入的特性,但对于染色质而言应同样表现出偏好转座的特点,即优先在开放染色质区插入[24].由此他们开展了实验验证,并获得了预期结果,从而建立了ATAC测序技术[20].
相较于DHs测序,ATAT测序具有难以比拟的优势,主要包括以下3点:(1)操作简单快捷.Tn5系统可以在转座打断DNA的同时将测序所需的接头连接到序列的端部,且操作十分简单快速,而这一优势同样在开放染色质检测方法中得以继承.如在获得细胞核后,ATAT测序只需要两个步骤,就可以构建出测序所需要的文库.其中,转座反应时间为5 min,加上后续纯化以及PCR富集等步骤,用时不超过3 h(图2);而DHs测序往往需要几十个实验处理步骤,其中包含两个过夜处理,完成整个流程至少需要3 d[20].(2)检测灵敏度提高.实验证明,ATAC测序在只有5万个细胞核起始量的情况下获得的DHs与DHs测序的结果具有很高的一致性,同时具有高于FAIRE测序的信噪比,而后两种方法所需要的细胞核起始量是ATAC测序的3~5个数量级.(3)实验重复性好.尤为重要的是,ATAC测序在技术重复间表现出非常好的可重复性(R=0.98),并与DHs测序数据间也有着较好的一致性(R>0.79)[20].并且,得益于改良的Tn5转座子的高频转座效率,ATAC测序也可以获得高分辨率的开放染色体区图谱.因此,ATAC测序方法与DHs测序方法一样,也被用于绘制核小体定位图谱及转录因子印记图谱.另外,ATAC测序无可比拟的样品需求量少、简便快速等特点也使得其应用在医学诊断上成为可能.因此,这一方法也得到了极大的关注,并因此被广泛应用[25-29].
1.4 THS测序
ATAC测序虽然具有简单快捷的特点,但是其在某些实验技术环节上仍存在不足之处.首先,由于Tn5通过插入剪断DNA并将测序接头连接到剪断的两个DNA片段的末端,因此对于一个DNA片段而言,其两端的接头连接是随机的,这便导致同一片段两端的接头有50%的概率是同一接头.而只有连接不同接头的片段才可用于富集扩增及测序,因此,有一半的片段无法利用.且大量剪断的DNA由于片段过大,无法进行PCR富集;再者,反应溶液的组成及反应条件方面仍然需要优化以便提高Tn5的活性及剪切效果.鉴于此,加州大学的一个研究团队在这些方面进行了探索,提出了一个新的技术方法,即THS测序.
相较于ATAC测序,这一方法在Tn5剪断DNA后在DNA末端加入了T7启动子,然后通过体外转录过程生成RNA片段,并通过RNA提取、cDNA合成以及PCR富集等类似于RNA测序的方法完成建库并获得数据.其改进之处就在于在转座子末端引入了T7启动子序列,转座剪断DNA后在DNA末端加上的是T7启动子序列,通过单链RNA生成来避免ATAC测序方法中存在的由于接头连接而导致的一半片段无法扩增利用的问题,同时也解决了大片段无法富集的问题.此外,研究团队也对转座酶进行了筛选,获得了活性更高的突变体,同时还对反应溶液及条件进行了优化,获得了更高效的转座效果.
采用这一方法对来自相同组织的100个细胞进行分析,结果发现所获得的开放染色质图谱与人类百科全书计划及杜克大学采用DHs测序获得的数据分别有61%和70%的重叠.同样,与采用5万个细胞的ATAT测序数据比较发现,它们之间的一致性达到61%[30].这也预示着,THS测序的检测敏感度较ATAT测序提高了500倍.进一步分析发现,采用THS测序获得的开放染色质位点数量是ATAC测序的1.5~2倍,证实其较ATAC测序具有更高检测敏感性.有趣的是,比较相同样品相同转座酶获得的数据发现,THS测序有超过25%的开放染色质位点的大小小于ATAC测序.这一点也说明ATAC测序存在一种偏好,即更容易检测区间较大的开放染色质位点,反之,相对小的位点则可能被漏检,导致数据完整性降低,而THS测序则可克服上述不足,数据更为完整可靠.
2 开放染色质的功能元件的注释分析
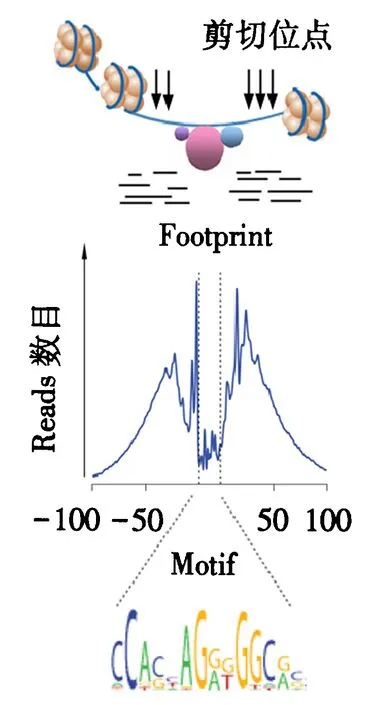
图3 调控因子印记分析Fig.3 Analysis of regulatory factor footprint
从原理上来看,开放染色质位点只是由于转录因子的结合导致的一个无核小体结构的裸露染色质区间,因此,无法从其定位来揭示具体的顺式元件及调控因子以及它们的功能等内在属性.因此,需要结合开放染色质位点的不同特征进行其功能元件的属性识别与鉴定.
2.1 调控因子印记
调控蛋白的结合导致其两侧染色质松散而形成DNase Ⅰ高度敏感区,与此相对应,调控蛋白结合的位置阻止了核酸酶、超声及转座子的剪切.因此,在保证作图精度的前提下,鉴定的开放染色质区内部应有一个不敏感区,它可以精确反应与调控因子互作的DNA序列,被称为调控因子的印记.通过调控因子印记分析可以辨识出与转录因子互作的保守域也就是顺式元件序列[31-32](图3).
就上述方法而言,DHs、ATAC和THS测序由于检测精度高,均可达到其要求,实现调控因子的印记分析.如在酵母中,研究人员通过分析发现了大量的DNase Ⅰ剪切的非敏感区,鉴定出4 384个调控因子印记,通过分析获得其保守域的序列,与已知的不同调控因子保守域序列比较发现,获得了近两个数量级的新的调控因子保守域[31].并且,研究人员发现,采用这一方法获得的保守域序列具有相对复杂的可塑性序列模式,如一个MADS box调控因子Mcm1的保守域中出现了3个小的受保护印记,而Cbf1位点则只有一个较大的印记.分析发现,这主要是由于不同类的调控因子与DNA具有不同的结合方式导致的[31](图4).这一现象在人类及拟南芥的研究中同样被发现并印证[32-33].这些发现对于揭示调控因子的属性及作用方式都有着重要的意义.
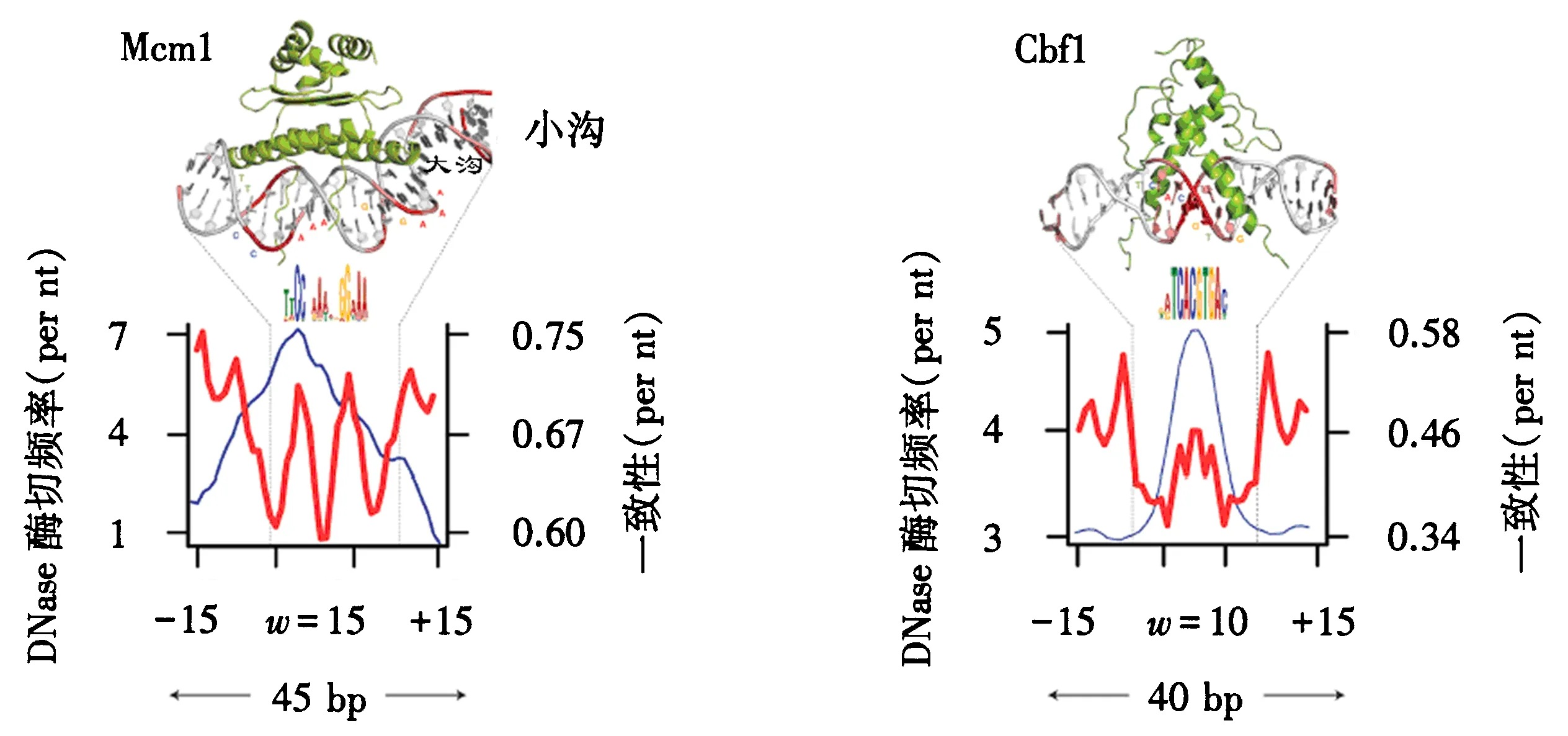
图4 调控因子Mcm1和Cbf1的印记分析Fig.4 Footprint analysis of regulatory factor Mcm1 and Cbf1
2.2 开放染色质位点与表观遗传学标记
表观遗传是指在DNA序列未发生改变的前提下发生的可遗传变异,是一种重要的遗传机制.研究发现,染色质上不同的功能区域往往具有不同的表观遗传特征[34].如在哺乳动物中,H3K4甲基化与乙酰化往往被认为与转录的启动有关,因此在启动子及转录起始位点区通常具有较高水平的H3K4me3和H3K4ac修饰[35-36];而增强子则往往具有较高水平的H3K4me1、H3K9me1、H3K27me1和H3K27ac修饰[37-38].虽然它们的内在机理仍不清楚,但这些表观特征为调控因子的鉴定提供了重要的线索和依据,尤其对于像增强子这类在序列组成及定位上均缺少共性的调控因子尤为重要.依据不同的表观修饰标记,研究人员从人类DHs中鉴定出约58万个启动子[14]及大量具有增强子活性的调控元件[11].在针对增强子的发掘分析中,Hnisz et al[39]利用DHs结合表观标记,从86种人类不同组织和细胞系中鉴定出200多个超强增强子.而对于单一基因而言,这一方法可以带来更为精确丰富的信息.如分析老鼠神经系统的一个重要基因Nefl的DHs测序数据发现,该基因区域存在多个DHs;结合不同组蛋白修饰数据发现不同的DHs表现出启动子、增强子等不同的修饰特征,而多种调控因子的存在也暗示该基因在表达调控上的复杂性[40](图5).在植物中,通过结合增强子的组蛋白修饰数据,同样发现在一个625 bp的DNA片段上存在着两个紧邻的增强子,有意思的是,它们分别在根和叶片中特异表达[41].由此可见,表观修饰在对DHs功能注释中具有重要作用.
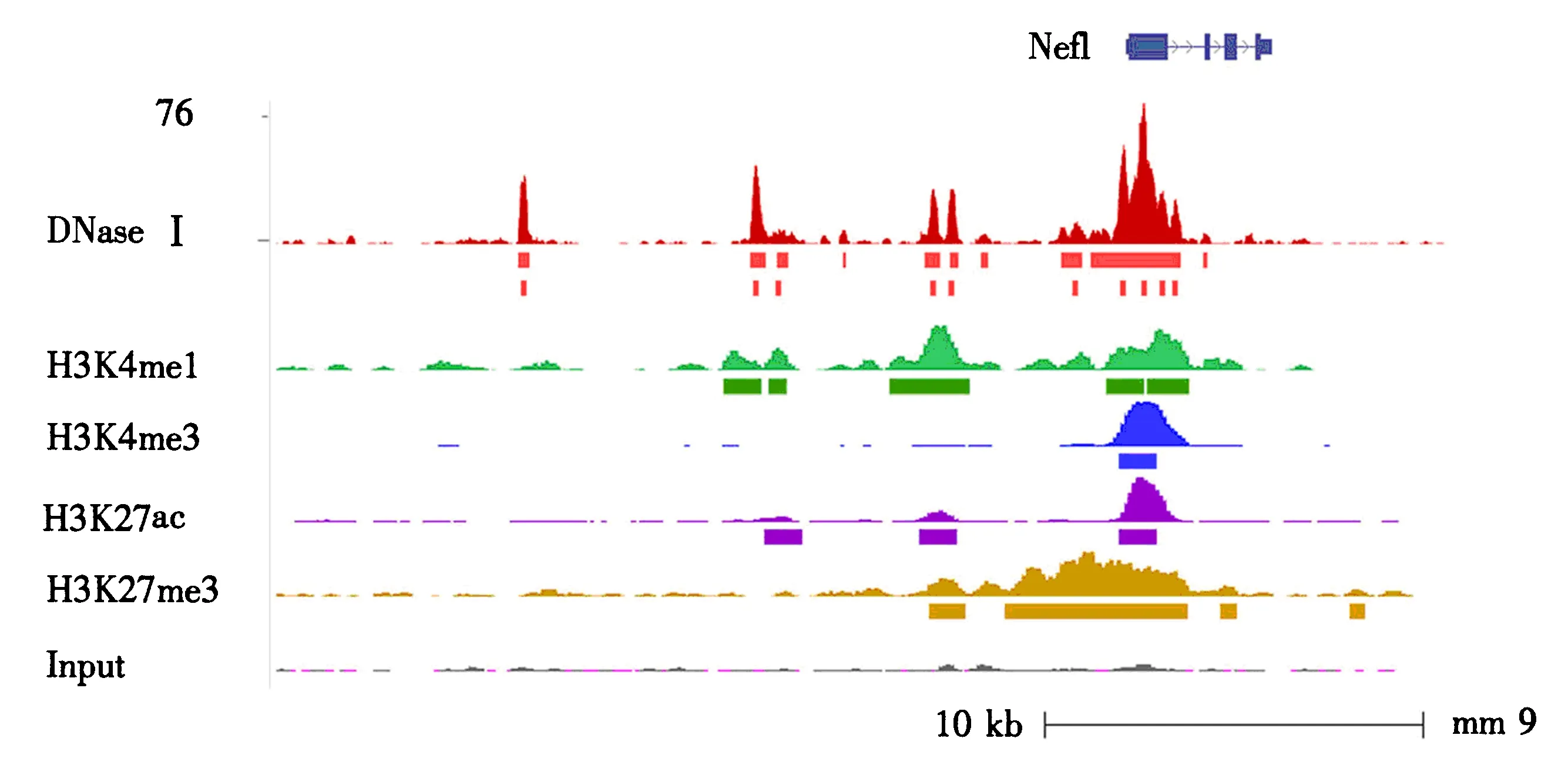
图5 DHs与表观遗传学修饰之间的关系Fig.5 Relation of DHs and epigenetic modification
2.3 采用染色质免疫沉淀进行开放染色质位点功能的注释
正如组蛋白修饰研究一样,通过特定调控因子抗体的染色质免疫沉淀技术可以分析出不同调控因子,如Pol Ⅱ、MYC和CTCF等的基因组结合位点,并与开放染色质位点比较分析,从而直接确定其调控因子的属性与功能.如通过比较CTCF的染色质免疫沉淀测序数据和DHs测序数据,人们发现并揭示了近30%的DHs是CTCF调控因子的作用位点[33];结合单细胞水平分析,研究者通过比较患病细胞的DHs测序数据与肿瘤抑制子p53的染色质免疫沉淀测序数据,发现了一个新的p53结合位点,并揭示这一结合位点的突变可以影响p53的绑定从而影响目标基因TXNL1的表达,而这极有可能是导致相关肿瘤产生的原因[15].但这一方法的前提是获得目标调控因子的抗体,因此只能针对已知调控因子进行研究,对于基因组中众多未知的调控因子,仍然需结合上述表观修饰和印迹分析等手段进行预测,并经实验验证获知其功能.
3 开放染色质位点在植物中的研究
植物中开放染色质的研究早有开展,但均局限于传统的Southern杂交方法.早在上世纪80年代,Spiker et al[42]采用DNase I酶切方法,首先揭示了DHs同样存在于小麦的基因区.之后,在大麦[43]、番茄[44]和拟南芥[45-46]上也有相关研究报道.但受传统的Southern杂交方法的限制,对植物DHs的研究也只限于以上物种.最近,美国威斯康辛大学的Jiming Jiang实验室将DHs测序引入植物DHs分析,从水稻[47]和拟南芥[32]不同组织中分别鉴定出约15万和4万个DHs.比较发现,在拟南芥一个80 kb区域,新方法不但鉴定出了几乎所有传统方法确定的DHs,同时还发现了大量新的DHs[32,46],证实了这一方法在植物中同样具有高效、可靠的特点.与其他物种结果相似,水稻和拟南芥中绝大多数高表达基因都与DHs相关联.与表观标记结合,研究者从拟南芥的DHs中预测出10 044个,可能为增强子,并从中选取了14个进行了功能验证.结果显示,10个为增强子[41],显示了这一方法在基因调控元件鉴定中的巨大潜力.最近,我国科研工作者采用DHs测序绘制了番茄开花后20 d及破色期果实的DHs图谱,并对该阶段特异发育基因的调控因子印记进行了分析,从而预测并揭示了与果实成熟有关的特定代谢途径上的调控因子[48].
4 展望
目前,随着基因组测序技术的发展,越来越多植物基因组测序完成,为功能基因组研究提供了极大的帮助.众所周知,作为功能基因组研究的主要对象之一的调控因子在基因组中不但数量众多,而且往往具有组织特异性,加之序列组成缺少共性,因此,难以采用比较基因组学的方法加以分析.如在基因表达调控网络中起到重要作用的增强子,其可以位于基因的上游、下游、内部,甚至与目标基因在不同的染色体上[49-51].而基于高通量测序的开放染色质位点发掘分析可以在基因组水平上揭示不同的调控因子位点,打破了单个基因上下游及染色体间的分析界线,为全面揭示基因表达调控网络提供了一个有力工具.
从技术方法原理可知,开放染色质并不能确定其真正的功能,因此仍应开展相应功能元件特征分析的工作,如各种组蛋白修饰、不同类型调控元件结构域分析以及基因组的三维构象模型分析,对开放染色质位点图谱进行功能注释.相信这将全面推动功能基因组的发展,为揭示生命功能机制开辟新的技术领域.
目前,就植物而言,开放染色质的研究十分有限,仅在模式材料水稻、拟南芥、番茄、玉米和二穗短柄草[32,41,47-48,52-53]上有所开展,且所用的是早期基于DNase Ⅰ或MNase酶切建立起来的方法;但值得思考的是,相对于动物较为简单的基因组,植物基因组多倍化现象普遍,DHs测序产生的短数据对这一类复杂的基因组来说,其应用是一个很大的挑战.而从技术方法上看,基于转座子的ATAC和THS测序使得上述困难得以解决,因而应重视开展植物相关的研究论证,希望本文的论述能为植物中相关研究的开展起到一定的推动作用.
[1] JIANG C, PUGH B F. Nucleosome positioning and gene regulation: advances through genomics [J]. Nat Rev Genet, 2009,10(3):161-172.
[2] MATHELIER A, SHI W, WASSERMAN W W. Identification of altered cis-regulatory elements in human disease [J]. Trends Genet, 2015,31(2):67-76.
[3] VARSHAVSKY A J, SUNDIN O H, BOHN M J. SV40 viral minichromosome: preferential exposure of the origin of replication as probed by restriction endonucleases [J]. Nucleic Acids Res, 1978,5(10):3 469-3 478.
[4] SCOTT W A, WIGMORE D J. Sites in simian virus 40 chromatin which are preferentially cleaved by endonucleases [J]. Cell, 1978,15(4):1 511-1 518.
[5] WU C, BINGHAM P M, LIVAK K J, et al. The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence [J]. Cell, 1979,16(4):797-806.
[6] STALDER J, LARSEN A, ENGEL J D, et al. Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNAase I [J]. Cell, 1980,20(2):451-460.
[7] WU C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I [J]. Nature, 1980,286(5 776):854-860.
[8] SUNDIN O, VARSHAVSKY A. Staphylococcal nuclease makes a single non-random cut in the simian virus 40 viral minichromosome [J]. J Mol Biol, 1979,132(3):535-546.
[9] KEENE M A, CORCES V, LOWENHAUPT K, et al. DNase I hypersensitive sites inDrosophilachromatin occur at the 5′ ends of regions of transcription [J]. Proc Natl Acad Sci USA, 1981,78(1):143-146.
[10] GROSS D S, GARRARD W T. Nuclease hypersensitive sites in chromatin [J]. Annu Rev Biochem, 1988,57:159-197.
[11] THURMAN R E, RYNES E, HUMBERT R, et al. The accessible chromatin landscape of the human genome [J]. Nature, 2012,489(7 414):75-82.
[12] CRAWFORD G E. Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites [J]. Proc Natl Acad Sci USA, 2004,101(4):992-997.
[13] SABO P J, HUMBERT R, HAWRYLYCZ M, et al. Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries [J]. Proc Natl Acad Sci USA, 2004,101(13):4 537-4 542.
[14] BOYLE A P, DAVIS S, SHULHA H P, et al. High-resolution mapping and characterization of open chromatin across the genome [J]. Cell, 2008,132(2):311-322.
[15] JIN W, TANG Q, WAN M, et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples [J]. Nature, 2015,528(7 580):142-146.
[16] LU F, LIU Y, INOUE A, et al. Establishing chromatin regulatory landscape during mouse preimplantation development [J]. Cell, 2016,165(6):1 375-1 388.
[17] GIRESI P G, KIM J, MCDANIELL R M, et al. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin [J]. Genome Res, 2007,17(6):877-885.
[18] SONG L, ZHANG Z, GRASFEDER L L, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity [J]. Genome Res, 2011,21(10):1 757-1 767.
[19] BEHURA S K, SARRO J, LI P, et al. High-throughput cis-regulatory element discovery in the vector mosquitoAedesaegypti[J]. BMC Genomics, 2016,17(1):1-11.
[20] BUENROSTRO J D, GIRESI P G, ZABA L C, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position [J]. Nat Methods, 2013,10(12):1 213-1 218.
[21] REZNIKOFF W S. Tn5 as a model for understanding DNA transposition [J]. Mol Microbiol, 2003,47(5):1 199-1 206.
[22] POTT S, LIEB J D. Single-cell ATAC-seq: strength in numbers [J]. Genome Biology, 2015, 16(1):1-4.
[23] CARUCCIO N. Preparation of next-generation sequencing libraries using NexteraTMtechnology: simultaneous DNA fragmentation and adaptor tagging by in vitro transposition [J]. Methods in Molecular Biology, 2011,733:241-255.
[24] GANGADHARAN S, MULARONI L, FAIN-THORNTON J, et al. DNA transposon Hermes inserts into DNA in nucleosome-free regionsinvivo[J]. Proc Natl Acad Sci USA, 2010,107(51):21 966-21 972.
[25] SCHARER C D, BLALOCK E L, BARWICK B G, et al. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naive SLE B cells [J]. Sci Rep, 2016,6:27 030.
[26] MILANI P, ESCALANTE-CHONG R, SHELLEY B C, et al. Cell freezing protocol suitable for ATAC-Seq on motor neurons derived from human induced pluripotent stem cells [J]. Sci Rep, 2016,6:25 474.
[27] WU J, HUANG B, CHEN H, et al. The landscape of accessible chromatin in mammalian preimplantation embryos [J]. Nature, 2016,534(7 609):652-657.
[28] ACKERMANN A M, WANG Z, SCHUG J, et al. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes [J]. Mol Metab, 2016,5(3):233-244.
[29] DAVIE K, JACOBS J, ATKINS M, et al. Discovery of transcription factors and regulatory regions drivinginvivotumor development by ATAC-seq and FAIRE-seq open chromatin profiling [J]. PLoS Genet, 2015,11(2):e1004994.
[30] SOS B C, FUNG H L, GAO D R, et al. Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay [J]. Genome Biol, 2016,17(1):20.
[31] HESSELBERTH J R, CHEN X, ZHANG Z, et al. Global mapping of protein-DNA interactionsinvivoby digital genomic footprinting [J]. Nat Meth, 2009,6(4):283-289.
[32] ZHANG W, ZHANG T, WU Y, et al. Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin inArabidopsis[J]. Plant Cell, 2012,24(7):2 719-2 731.
[33] BOYLE A P, SONG L, LEE B K, et al. High-resolution genome-wideinvivofootprinting of diverse transcription factors in human cells [J]. Genome Res, 2011,21(3):456-464.
[34] BLACK JOSHUA C, VAN RECHEM C, WHETSTINE JOHNATHAN R. Histone lysine methylation dynamics: establishment, regulation, and biological impact [J]. Mol Cell, 2012,48(4):491-507.
[35] BARSKI A, CUDDAPAH S, CUI K, et al. High-resolution profiling of histone methylations in the human genome [J]. Cell, 2007,129(4):823-837.
[36] WANG Z, ZANG C, ROSENFELD J A, et al. Combinatorial patterns of histone acetylations and methylations in the human genome [J]. Nat Genet, 2008,40(7):897-903.
[37] RADA-IGLESIAS A, BAJPAI R, SWIGUT T, et al. A unique chromatin signature uncovers early developmental enhancers in humans [J]. Nature, 2011,470(7 333):279-283.
[38] ONG C T, CORCES V G. Enhancers: emerging roles in cell fate specification [J]. EMBO Rep, 2012,13(5):423-430.
[39] HNISZ D, ABRAHAM BRIAN J, LEE TONG I, et al. Super-enhancers in the control of cell identity and disease [J]. Cell, 2013,155(4):934-947.
[40] WILKEN M, BRZEZINSKI J, LA TORRE A, et al. DNase I hypersensitivity analysis of the mouse brain and retina identifies region-specific regulatory elements [J]. Epigenetics & Chromatin, 2015,8(1):8.
[41] ZHU B, ZHANG W, ZHANG T, et al. Genome-wide prediction and validation of intergenic enhancers inArabidopsisusing open chromatin signatures [J]. Plant Cell, 2015,27(9):2 415-2 426.
[42] SPIKER S, MURRAY M G, THOMPSON W F. DNase I sensitivity of transcriptionally active genes in intact nuclei and isolated chromatin of plants [J]. Proc Natl Acad Sci USA, 1983,80(3):815-819.
[43] STEINM LLER K, BATSCHAUER A, Apel K. Tissue-specific and light-dependent changes of chromatin organization in barley (Hordeumvulgare) [J]. Eur J Biochem, 1986,158(3):519-525.
[44] CONCONI A, RYAN C A. DNase I and micrococcal nuclease analysis of the tomato proteinase inhibitor I gene in chromatin [J]. J Biol Chem, 1993,268(1):430-435.
[45] VEGA-PALAS M A, FERL R J. TheArabidopsisAdhgene exhibits diverse nucleosome arrangements within a small DNase I-sensitive domain [J]. Plant Cell, 1995,7(11):1 923-1 932.
[46] KODAMA Y, NAGAYA S, SHINMYO A, et al. Mapping and characterization of DNase I hypersensitive sites inArabidopsischromatin [J]. Plant Cell Physiol, 2007,48(3):459-470.
[47] ZHANG W, WU Y, SCHNABLE J C, et al. High-resolution mapping of open chromatin in the rice genome [J]. Genome Res, 2012,22(1):151-162.
[48] QIU Z, LI R, ZHANG S, et al. Identification of regulatory DNA elements using genome-wide mapping of DNase I hypersensitive sites during tomato fruit development [J]. Mol Plant, 2016,9(8):1 168-1 182.
[49] CUI L, MURCHLAND I, SHEARWIN K E, et al. Enhancer-like long-range transcriptional activation by λ CI-mediated DNA looping [J]. Proc Natl Acad Sci USA, 2013,110(8):2 922-2 927.
[50] VAHEDI G, TAKAHASHI H, NAKAYAMADA S, et al. STATs shape the active enhancer landscape of T cell populations [J]. Cell, 2012,151(5):981-993.
[51] VAHEDI G, KANNO Y, FURUMOTO Y, et al. Super-enhancers delineate disease-associated regulatory nodes in T cells [J]. Nature, 2015,520(7 548):558-562.
[52] ZHANG T, Marand A P, JIANG J. PlantDHS: a database for DNase I hypersensitive sites in plants [J]. Nucleic Acids Res, 2015,44(D1):1 148-1 153.
[53] RODGERSMELNICK E, VERA D L, BASS H W, et al. Open chromatin reveals the functional maize genome. [J]. Proc Natl Acad Sci USA , 2016,113(22):3 177-3 184.
(责任编辑:施晓棠)
Progress on genome-wide identification and analysis of transcriptional regulatory elements based on open-chromatin signatures
HAN Jinlei1,2, LI Zhanjie1, WANG Kai1,2,3
(1.Center for Genomics and Biotechnology, Haixia Institute of Science and Technology; 2.College of Crop Science; 3.National Engineering Research Center of Sugarcane, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China)
Eukaryote′s gene expression and regulation are the consequence of interactions between cis-acting elements and trans-acting factors. It is known that these interaction processes are associated with the dynamics of nucleosome positioning, and the binding of regulatory factors that require naked DNA region, which is an open chromatin site. Thus, the accurate mapping of open chromatin sites can provide an effective solution to identify regulatory elements, and even uncover gene regulatory mechanism. In this paper, we review the definition, main research methods and functional annotation of open chromatin sites, with an aim of providing references for discovering regulatory elements in genome level, especially in plant.
open-chromatin site; DNase Ⅰ hypersensitive site sequencing; regulator footprint; epigenetic modification
2016-07-07
2016-11-09
国家自然科学基金资助项目(31471170、31628013);福建农林大学人才引进启动项目(11899006004);棉花生物学国家重点实验室开放课题(CB2015A05).
韩金磊(1989-),男,硕士研究生.研究方向:作物遗传育种. Email:1140102020@fafu.edu.cn.通讯作者王凯(1977-),男,教授,博士生导师.研究方向:作物遗传育种. Email:kwang@fafu.edu.cn.
Q756
A
1671-5470(2017)01-0001-08
10.13323/j.cnki.j.fafu(nat.sci.).2017.01.001
