密闭微波辅助提取-HPLC法测定还亮草中硬飞燕草碱和巴比翠雀碱的含量
2017-02-14樊轻亚郝艳红代春美
樊轻亚,郝艳红,代春美
(1.信阳职业技术学院 药学院, 河南 信阳 464000;2.信阳职业技术学院 医学院, 河南 信阳 464000;3.中国中医科学院,北京 100700)
密闭微波辅助提取-HPLC法测定还亮草中硬飞燕草碱和巴比翠雀碱的含量
樊轻亚1*,郝艳红2,代春美3
(1.信阳职业技术学院 药学院, 河南 信阳 464000;2.信阳职业技术学院 医学院, 河南 信阳 464000;3.中国中医科学院,北京 100700)
采用密闭微波辅助法(PMAE)提取还亮草中的硬飞燕草碱和巴比翠雀碱。采用单因素试验结合正交试验方法对微波实验条件进行优化。得到硬飞燕草碱和巴比翠雀碱的最佳提取方案:药物颗粒度100目,固液比1∶60,微波温度80 ℃,微波功率560 W,微波时间10 min。以甲醇-0.2%三乙胺(45∶55)为流动相,建立了高效液相色谱(HPLC)测定硬飞燕草碱和巴比翠雀碱含量的方法。硬飞燕草碱和巴比翠雀碱分别在0.50~50.0,0.30~30.0 mg/L范围内呈良好的线性关系,回收率为98.3%~104.5%,相对标准偏差(RSD)分别为2.0%和2.2%。与传统溶剂回流法(SRE)进行比较,该方法简单、提取率更高。
还亮草;密闭微波提取;硬飞燕草碱;巴比翠雀碱;正交试验
还亮草(Delphinium anthriscifolium Hance)是毛茛科、翠雀属多年生草本植物,分布于中国广东、广西、贵州、湖南、江西等地,有祛风除湿、止痛活络功效,用于治疗风湿痛、半身不遂、食积胀满、咳嗽;外用于痈疮癣疥[1]。药用还亮草主要含有生物碱成分,另外还有固定油、类脂、苷类等成分。硬飞燕草碱和巴比翠雀碱是还亮草中含量较多的生物碱类成分。研究发现硬飞燕草碱有箭毒样作用,并有神经节阻断作用,可用于肌紧张或运动过多症,巴比翠雀碱则具有解痉作用,是治疗急性菌痢、肠炎的有效成分[1]。
微波辅助提取技术以其加热迅速、耗能低、操作时间短、溶剂耗量少、选择性好、提取效率高等优点受到广泛重视[2-17],目前主要有密闭式微波提取系统(PMAE)和聚焦微波提取系统(FMAE)。PMAE提取法是利用微波对置于密闭提取罐中的药材和提取溶剂进行照射,使药材在高温高压的环境下被提取,可同时处理多个样品,提取快速、高效;FMAE是在常压微波辐射下进行开放式提取,安全、样品处理量大,但设备需改装,受穿透能力限制,提取不均匀,提取时间长,且易导致挥发性物质损失。本文首次采用PMAE-高效液相色谱法测定还亮草中硬飞燕草碱和巴比翠雀碱的含量,通过单因素实验对颗粒度、固液比、微波温度、微波功率和微波时间等条件进行考察,进一步通过正交试验的优化得到最佳提取工艺,大大增加了提取率。该法避免了使用大量有机溶剂,不破坏环境,降低了污染,是生物碱类化合物有效的提取方法。
1 实验部分
1.1 试验药材、试剂与仪器
巴比翠雀碱标准品、硬飞燕草碱标准品(成都普菲德生物技术有限公司,纯度>98%),还亮草药材(产地:广西),甲醇、乙腈(色谱纯,美国Merck 公司),三乙胺、氨水、95%乙醇、盐酸、氯仿(分析纯,郑州德众化学试剂厂),实验用水为蒸馏水。
Agilent 1200高效液相色谱仪(美国安捷伦公司);B-260恒温水浴锅(上海亚荣生化仪器厂);SHB-Ⅲ循环水式多用真空泵(郑州长城科贸有限公司);RE52CS-1旋转蒸发仪(上海亚荣生化仪器厂);KQ2200B超声波清洗仪(昆山市超声仪器有限公司);TOPEX全能型微波化学工作平台(上海屹尧微波化学有限公司)。
1.2 实验方法
1.2.1 PMAE提取称取一定量的还亮草药材(100目)于提取罐中,按照1∶60的固液比加入95%乙醇后,置于密闭微波瓶中加热提取(10 min,80 ℃),提取液冷却至室温。
1.2.2 SRE提取称取一定量的100目还亮草药材于提取罐中,按照1∶60的固液比加入95%乙醇后置于水浴上(80 ℃,提取40 min),提取液冷却至室温。
1.2.3 提取液的纯化将提取液减压抽滤,滤液旋干,用30 mL盐酸溶液(pH 2.0)超声溶解,倾出,置于分液漏斗,氯仿萃取后的水相用1%NaOH溶液调至pH 10.0,再用氯仿萃取3次,每次30 mL,收集氯仿层,旋干,用甲醇溶解、定容,HPLC含量待测。
1.2.4 HPLC测定条件色谱柱(Phenomenon Gemini C18柱,4.6 mm×250 mm,5 mm);流动相:甲醇-0.2%三乙胺(45∶55);检测波长:254 nm;流速:1 mL/min;柱温:室温;进样量:10 mL。
2 结果与讨论
2.1 单因素试验
2.1.1 药材颗粒度和固液比的影响样品颗粒度大小直接影响微波的穿透能力,从而影响微波加热的速率和均匀性。考察了药材颗粒度(70,80,90,100,110目)对提取率的影响,结果表明,提取率随着药材颗粒度的增大而增加,当样品颗粒度为100目时提取率最高,继续增大颗粒度时提取率反而降低。这是因为颗粒度太大时微波难以完全穿透样品,破壁不完全;颗粒度太小时微波虽能完全穿透样品,但由于相邻细胞间的相互作用增强,细胞所受阻力增大,因而其内部细胞被破坏的程度相对较小。而且实验发现颗粒度太小时,微波照射时易出现焦化,会影响有效成分的提取。因此选择样品的最佳颗粒度为100目。
实验还考察了固液比(1∶20~1∶60)对硬飞燕草碱及巴比翠雀碱的影响(图1A)。结果表明,固液比为1∶60时有效成分的提取率最大,而固液比在1∶50和1∶60时提取率相差不大,说明物料已达到饱和,这可能是因为有效成分在溶剂中的溶解度一定,增加固液比相当于增加了溶剂量,所以有效成分的溶解量会逐渐增加,直至达到饱和状态,之后提取量基本保持恒定。
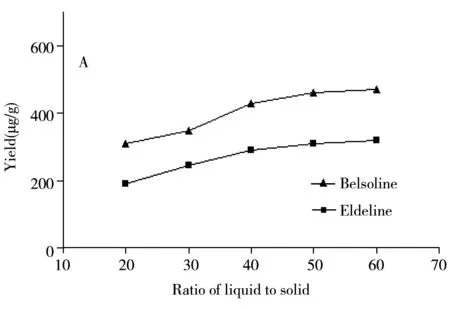
2.1.2 微波温度、微波功率、微波时间的影响由于微波温度较低不利于细胞膜内有效成分的释出,使之提取不完整,提取率降低,而过高的微波温度又会使待测物质不稳定,造成有效成分的降解,并且烧焦药材,提取率降低。本实验考察了微波温度(60~100 ℃)对硬飞燕草碱、巴比翠雀碱的影响,结果显示,在80 ℃时硬飞燕草碱和巴比翠雀碱达到最大提取率,温度过低或过高均会导致提取率降低。
考察了微波功率对两种有效成分提取率的影响(图1B)。结果表明,随着微波功率的增加,提取率升高,但功率过高时,样品因受热过度而焦化,颗粒粘结,使得生物碱类成分更难被提取出,因而造成提取率下降。实验选择微波温度80 ℃,微波功率560 W,此时可获得最佳提取率。
微波时间主要取决于大部分有效成分释放出的时间,时间过长,可能使已释出的有效成分降解,时间过短则释出不完全。实验考察了不同微波时间(2~18 min)对硬飞燕草碱和巴比翠雀碱的影响。结果显示,硬飞燕草碱和巴比翠雀碱在10 min时达到最大提取率,说明微波时间小于10 min时,有效成分未被完全提出,而微波时间大于10 min时,有效成分可能发生降解,导致提取率降低。因此选择最佳微波时间为10 min。

表1 硬飞燕草碱、巴比翠雀碱的正交试验因素水平表
2.2 正交试验优化萃取条件
根据单因素试验的结果,由于微波温度、微波功率、微波时间之间存在交互影响作用,所以选取微波温度、微波功率、微波时间3个因素,以硬飞燕草碱和巴比翠雀碱的提取率为指标,L9(34)正交试验设计见表1。实验结果见表2~3。
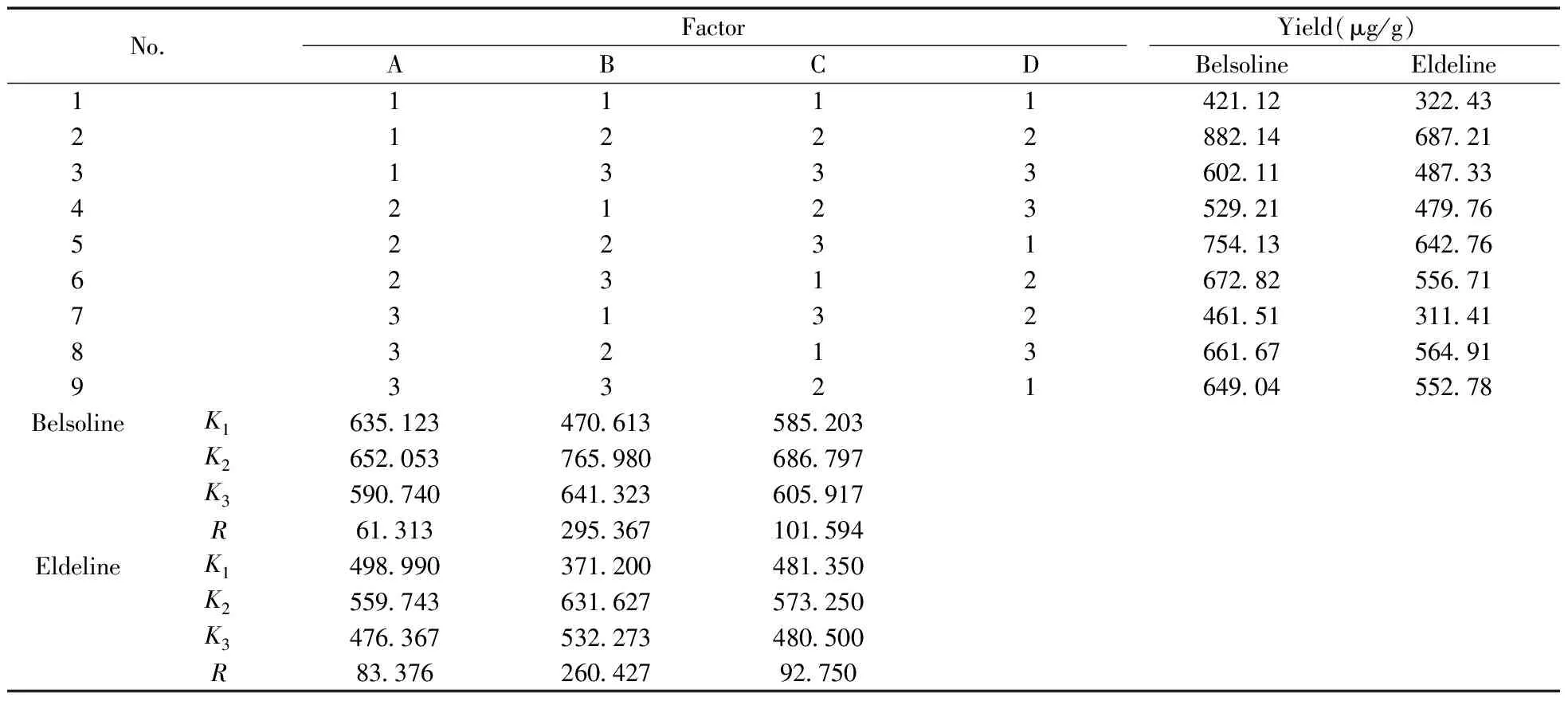
表2 正交试验设计及测定结果

表3 方差分析结果
由表2~3可见,在不同萃取条件下测得的还亮草中硬飞燕草碱、巴比翠雀碱的提取率相差很大,影响硬飞燕草碱和巴比翠雀碱提取率的因素主次顺序为:B>C>A,即微波功率>微波时间>微波温度,最佳萃取方案为A2B2C2,即微波温度80 ℃,微波功率560 W,微波时间10 min。这是因为当微波功率太低,辐射时间太短的情况下,产生的热量少,细胞中局部过热的现象不明显,因而破壁效果差,以致硬飞燕草碱和巴比翠雀碱的提取率较低;而微波辐射时间太长,微波功率太高,还亮草样品因受热过度而焦化,样品颗粒粘结,有效成分更难被提取出,因而造成提取率下降。
在最优条件下,取3份药材进行验证,结果表明按照最佳方案A2B2C2萃取得到硬飞燕草碱的提取率可达913.35 μg/g,巴比翠雀碱可达706.32 μg/g,因此采用最优条件A2B2C2提取还亮草中的硬飞燕草碱和巴比翠雀碱是可行的。
2.3 HPLC分析结果
采用HPLC法对硬飞燕草碱和巴比翠雀碱进行含量测定。分别取一定量的标准品及还亮草药材的提取液,按HPLC条件进行分离,色谱图如图2所示。
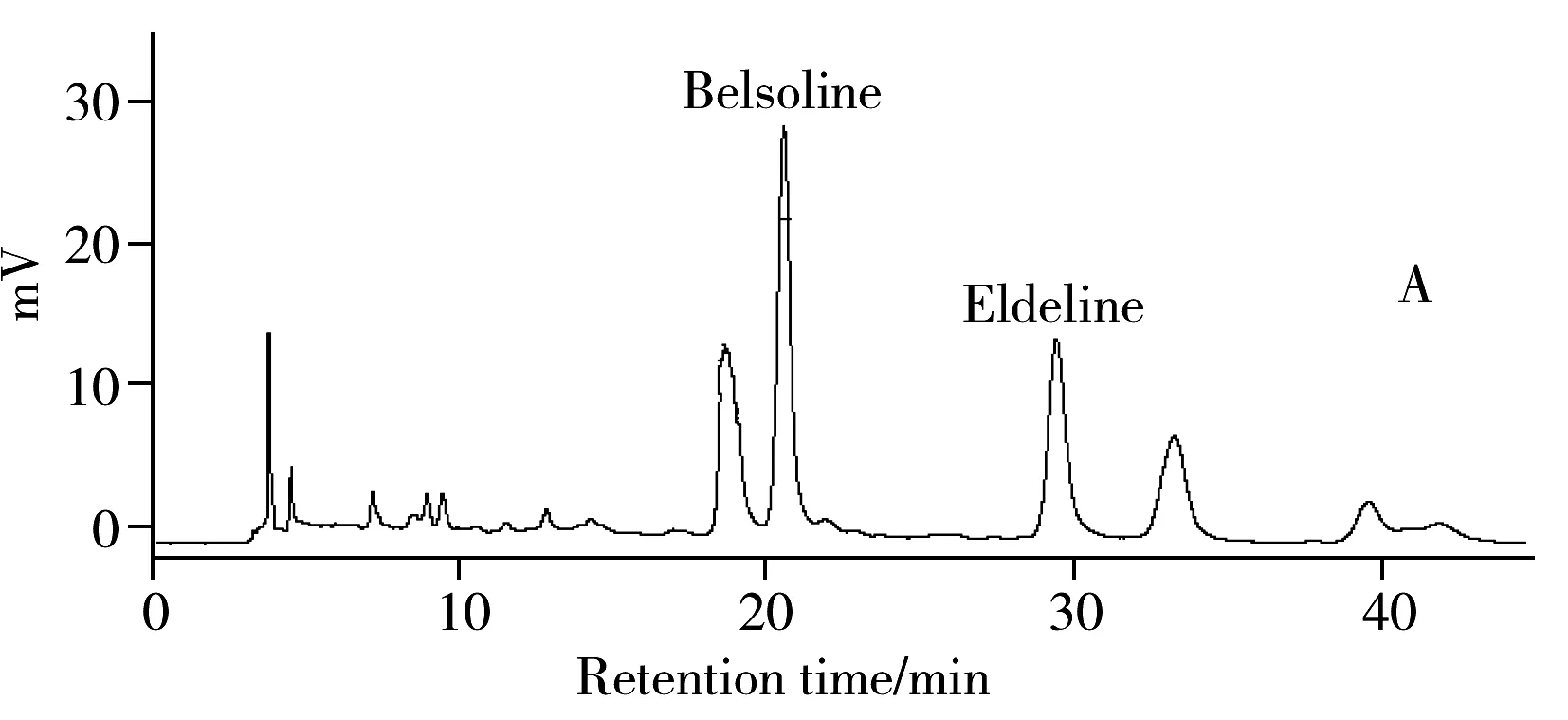
由图2可见,以甲醇-0.2%三乙胺(45∶55)作为流动相,在254 nm波长处检测,硬飞燕草碱和巴比翠雀碱可以得到良好的分离,分离度(R)大于1.5,保留时间分别为20.65 min和30.37 min。
2.3.1 线性关系与检出限取一定量的硬飞燕草碱和巴比翠雀碱混合标准溶液,分别配制成一系列浓度的硬飞燕草碱和巴比翠雀碱混合标准品,分别进样10 μL,以其峰面积(A)对浓度(C,mg/L)绘制标准曲线;按3倍信噪比(S/N=3)计算检出限,结果见表4。 由表4可知,硬飞燕草碱、巴比翠雀碱的线性范围分别为0.50~50.0,0.30~30.0 mg/L,相关系数均大于0.999,方法可满足还亮草提取液中生物碱的测定要求。

表4 硬飞燕草碱、巴比翠雀碱的标准工作曲线、线性范围与检出限
2.3.2 精密度、稳定性与重复性精密吸取供试品溶液10 μL,连续进样5次,测得硬飞燕草碱、巴比翠雀碱的相对标准偏差(RSD)分别为0.9%和1.2%,表明仪器精密度良好;分别在溶液配制2,4,6,8,10,12 h后进样,进行稳定性试验,测得硬飞燕草碱、巴比翠雀碱的RSD值分别为1.8%和1.2%,表明提取液中的硬飞燕草碱、巴比翠雀碱可在12 h内保持稳定。按本方法进行6次平行试验,得硬飞燕草碱、巴比翠雀碱的RSD值为1.7%和1.9%,表明方法的重复性良好。
2.3.3 加标回收试验分别准确称取药材5份,加入一定量的900 μg/mL硬飞燕草碱和700 μg/mL巴比翠雀碱混合标准品,按“1.2.1”方法提取、预处理,计算硬飞燕草碱、巴比翠雀碱的回收率,结果见表5。硬飞燕草碱、巴比翠雀碱的回收率为98.3%~104.5%,RSD分别为2.0%和2.2%,方法的准确度及精密度满足分析检测的要求。
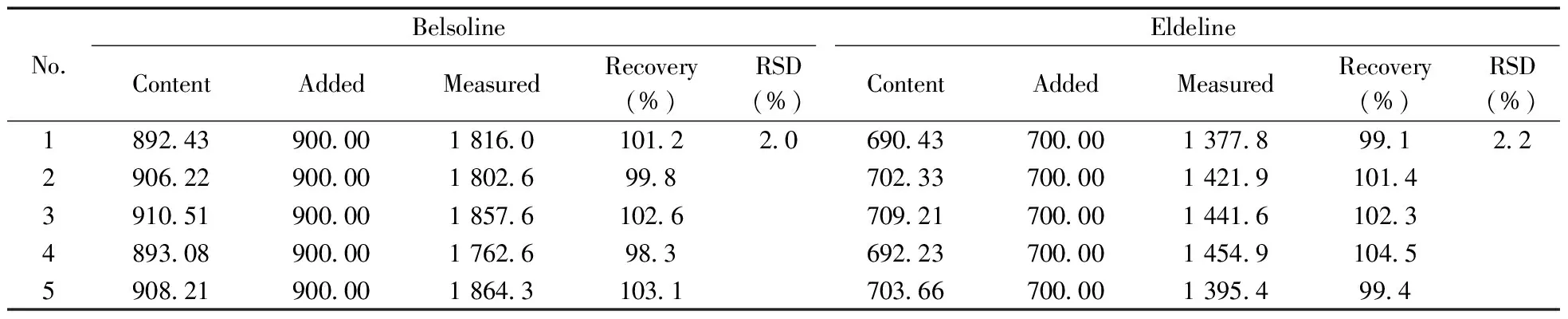
表5 硬飞燕草碱和巴比翠雀碱的加标回收率与相对标准偏差(n=3)
2.4 微波辅助提取法与溶剂回流提取法的比较
生物碱通常采用溶剂回流提取法(SRE)进行提取,因此本文采用SRE法的最佳提取方案“1.2.2”与密闭微波辅助提取法的最佳提取方法“1.2.1”对3批次还亮草进行比较研究,结果见表6。

表6 不同批次的提取分析结果(n=3)
由表6可知,采用SRE法提取还亮草中生物碱的提取率只有PMAE法的2/3,这表明不同的提取加热方式对生物碱的提取率影响较大。传统的SRE法在提取过程中首先是在大气环境中加热溶剂并溶胀样品细胞,以热传导方式将热能从溶剂传递到细胞内部进行加热。而PMAE法中95%乙醇是吸收微波的良好溶剂,与细胞内的极性物质共同吸收微波,并迅速转变为热能,提取溶剂及药材被迅速整体性的“体加热”,且在具有搅拌装置的密闭体系中,溶剂蒸汽有一定压力,溶剂回流效果及传热效率高,提取率大大提高。由于PMAE的均匀加热以及密闭微波装置的电磁搅拌克服了传统提取方式加热不均匀的缺点,改善了提取的重现性。因此采用PMAE提取还亮草中生物碱,快速、高效,提取效率优于传统SRE提取方法。
3 结 论
本文考察了密闭微波辅助提取高效液相色谱测定还亮草中硬飞燕草碱和巴比翠雀碱含量的方法。该方法操作便捷,精密度高,准确度好,并能保护被提取物质原有特性,能很好的提取出有效成分,避免使用大量有机溶剂,降低了污染,适应于还亮草中有效成分的提取分析,对其他中药材的分析具有一定的指导意义。
[1] Editorial Committee of Flora of Chinese.FloraChinese.Beijing:Science Press(《中国植物志》编辑委员会.中国植物志.北京:科学出版社),1979,27:460.
[2] Tan X Y.Guid.J.Tradit.Chin.Med.Pharm.(谭笑银.中医药导报),2013,19(2):101-103.
[3] Gu Z W,Hu T,Jia Y,Tan Y,Zhu X Y,Li J L.J.Chin.Med.Mater.(谷政伟,胡铁,贾媛,谭云,朱晓媛,黎继烈.中药材),2014,37(11):2092-2095.
[4] Zhu D,He S L,Fan H J,Wang L P,Lou X X,Wan Q,Zang L Q.J.Instrum.Anal.(朱丹,何世玲,范华均,王李平,娄欣欣,万强,臧林泉.分析测试学报), 2013,32(9):1075-1080.
[5] Fan Q Y,Li G X.J.Instrum.Anal.(樊轻亚,李光霞.分析测试学报),2015,34(5):605-609.
[6] Kong N,Zou X B,Huang R,Tao J Z,Wei X Y.J.Instrum.Anal.(孔娜,邹小兵,黄锐,陶进转,魏欣旸.分析测试学报),2010,29(10):1102-1108.
[7] Li G K,Du F Y,Xiao X H.J.Instrum.Anal.(李攻科,杜甫佑,肖小华.分析测试学报),2007,24(12):1184-1191.
[8] Wei S L,Liu J H,Yan Z J,Deng G H.J.Instrum.Anal.(韦寿莲,刘君红,严子军,邓光辉.分析测试学报),2009,28(7):773-779.
[9] Fan Q Y,Wu C Z,Wang W H.J.Instrum.Anal.(樊轻亚,吴长忠,王万好,分析测试学报),2014,33(9):1073-1077.
[10] Wang L P,Liu X Q,Fan H J,She X H,Xiao X X,Huang X W.Phys.Test.Chem.Anal.:Chem.Anal.(王李平,刘小琴,范华均,佘旭辉,肖雪霞,黄晓文.理化检验:化学分册),2013,49(2):129-133.
[11] Zhao C C,Cheng W,Wang L D.Chin.Tradit.HerbalDrugs(赵晨晨,承伟,王立冬.中草药),2012,43(4):718-720.
[12] Nie J Y,Wu C Y,Wu S R,Li Z L.Chin.Tradit.HerbalDrugs(聂金媛,吴成岩,吴世容,李志良.中草药),2004,35(12):1346-1348.
[13] Liu X R,Cai P P,Jiang T,Xiang X Z,Lü J Q.Chin.Tradit.PatentMed.(刘学仁,蔡品品,姜涛,向显智,吕鉴泉.中成药),2013,35(2):420-423.
[14] Fan Q Y,Fan H J,Huang X W,Huang Y,Zhang Y F.Chin.Tradit.PatentMed.(樊轻亚,范华均,黄晓文,黄勇,张煜帆.中成药),2010,32(2):326-329.
[15] Wang G H, Zhou F,Ren Z N,Yang Y.J.Anal.Sci.(王桂花,周峰,任中楠,杨屹.分析科学学报),2009,25(5):559-562.
[16] Chen M,Yuan D X, Xu P X.J.Instrum.Anal.(陈猛,袁东星,许鹏翔.分析测试学报),1999,18(2):82-86.[17] Song W F,Tao L,Zhong M.J.Instrum.Anal.(宋伟峰,陶玲,钟鸣.分析测试学报),2012,31(7):810-815.
Determination of Belsoline and Eldeline Contituents in Delphinium Anthriscifolium Hance by Microwave-assisted Extraction with HPLC
FAN Qing-ya1*, HAO Yan-hong2,DAI Chun-mei3
(1.College of Pharmacy,Vocational and Technical College School of Xinyang,Xinyang 464000,China;2.College of Medicine,Vocational and Technical College School of Xinyang,Xinyang 464000,China;3.China Academy of Chinese Medical Sciences,Beijing 100700,China)
A method of extraction of belsoline and eldeline in Delphinium anthriscifolium Hance was developed by pressurized microwave-assisted extraction(PMAE).The single factor test and orthogonal test were used to optimize the experimental conditions.The optimum extraction conditions were as follows:particle size:100 mesh,ratio of solid to liquid:1∶60,microwave temperature:80 ℃,microwave power:560 W,microwave time:10 min.Under the optimal conditions,belsoline and eldeline were separated,and determined by HPLC using methanol-0.2% triethylamine(45∶55) as mobile phase.The linear ranges of belsoline and eldeline were in the ranges of 0.50-50.0 mg/L and 0.30 -30.0 mg/L,respectively.The recoveries of belsoline and eldeline were in the range of 98.3%-104.5%,and RSDs were 2.0% and 2.2%,respectively.Compared with the solvent reflux extraction method(SRE) ,the proposed method was simple,and showed high extraction efficiencies.
Delphinium anthriscifolium Hance;pressurized microwave-assisted extraction;belsoline;eldeline;orthogonal test
10.3969/j.issn.1004-4957.2017.01.018
2016-07-20;
2016-08-21
第24批中国博士后科技服务团合作项目
*通讯作者:樊轻亚,硕士,讲师,研究方向:药物提取分离及分析, Tel:0376-6281969,E-mail:fqy2006hn@163.com
O657.72;TQ460.72
A
1004-4957(2017)01-0106-06
