反钙钛矿结构RE3AlC热物理性质理论研究
2017-01-18李德怀卢经文陈红梅陶小马欧阳义芳
李德怀,袁 文,卢经文,张 凡,陈红梅,陶小马,欧阳义芳
(广西大学物理科学与工程技术学院,广西 南宁 530004)
反钙钛矿结构RE3AlC热物理性质理论研究
李德怀,袁 文,卢经文,张 凡,陈红梅,陶小马,欧阳义芳†
(广西大学物理科学与工程技术学院,广西 南宁 530004)
结合第一性原理和准谐德拜模型计算了RE3AlC(RE=Sc、Y及镧系稀土)系列具有反钙钛矿结构碳化物的德拜温度、格律乃森常数、体积模量、自由能、比热容等热物理性质随着温度和压强变化的趋势。结果表明RE3AlC碳化物的比热容、体积模量以及吉布斯自由能等随温度和压强变化的总趋势相似,其中RE3AlC碳化物的体积弹性模量随着温度的升高而逐渐减小,同时随着压强的增加而增大;吉布斯自由能都随着温度的升高而降低,其中Sc3AlC化合物的自由能最低,而Yb3AlC化合物的自由能最高,表明Sc3AlC化合物最稳定,而Yb3AlC化合物稳定性最低;等容比热容随着温度和压强的变化在0-300 K温度段内变化较大,随后趋于平缓逐渐趋于杜隆-帕蒂极限值。
RE3AlC;德拜模型;热物理性质
1 引言
近几十年来,材料科学已经成为理化工程、环境保护和能源医药等诸多领域所关注的焦点。RE3AlC(RE=Sc、Y及镧系稀土)作为一种重要的具有反钙钛矿结构的碳化物,因其高杨氏模量、高熔点等与钙钛矿结构化合物相似的性质,使得其在电子工业、化工等方面拥有极大的潜在应用。目前对这类化合物的热物理性质,特别是RE3AlC的热物理性质至今没有文献报道。因此,本文利用第一性原理结合准谐徳拜模型的方法对具有反钙钛矿结构的碳化物RE3AlC的相关性能进行了研究,主要包括材料的德拜温度、格律乃森常数、体积模量、吉布斯自由能、比热容随温度和压强的变化关系,这将丰富具有反钙钛矿结构晶体的热物理性质,为其潜在应用提供参考。
2 计算方法
本文采用第一原理计算软件包Vienna Ab initio Simulation Package (VASP)[1]进行计算。计算方法是投影缀加波方法[2,3],交换关联势采用广义梯度近似的PBE方案[4]。布里渊区积分采Monkhorst Pack布点方法[5]。本文计算中截断能设置为600 eV,能量收敛精度为10-6eV/atom。计算结果误差小于1.0 meV/atom。通过第一性原理获得了能量与体积的关系,结合准谐近似的德拜模型获得了RE3AlC的热力学性质,有关具体的计算方法可参考文献[6-9]。
3 结果和讨论
3.1 晶格常数、德拜温度和格律乃森常数
本文计算的晶格常数、德拜温度、格律乃森常数值如表1所示。从表中可以看出,计算出的RE3AlC的晶格常数值和实验值[10]的结果较为一致,先升高后降低,误差较小说明本文计算合理。其中,Yb3AlC晶格常数的计算值比实验值[10]稍大,这主要可能是因为势函数中对f电子的处理有关,文献[10]中提到在Yb3AlC化合物中Yb应该是3价的,在本文计算中,选用3价或者2价的Yb势函数都不能获得与实验值相吻合的计算值,这需要从势函数和实验两方面进行进一步确认。表1中还给出了压强为0 GPa下RE3AlC的德拜温度和格律乃森常数的计算值,从表1可以看出,RE3AlC在温度为300 K下的德拜温度值要低于0K下的德拜温度值,说明随着温度的升高,RE3AlC的德拜温度是降低的;另外,在同温度段下(如T=0K),Sc3AlC的德拜温度明显高于其它化合物,Yb3AlC的德拜温度值要明显低于其它化合物;对于镧系元素,除了Yb3AlC,其RE3AlC的德拜温度实随着原子序数的增加而增加的。一般来说,德拜温度越高,化合物的结合越强。从格律乃森常数值可以发现,Sc3AlC的格律乃森常数值最高,Yb3AlC的格律乃森常数值最低。
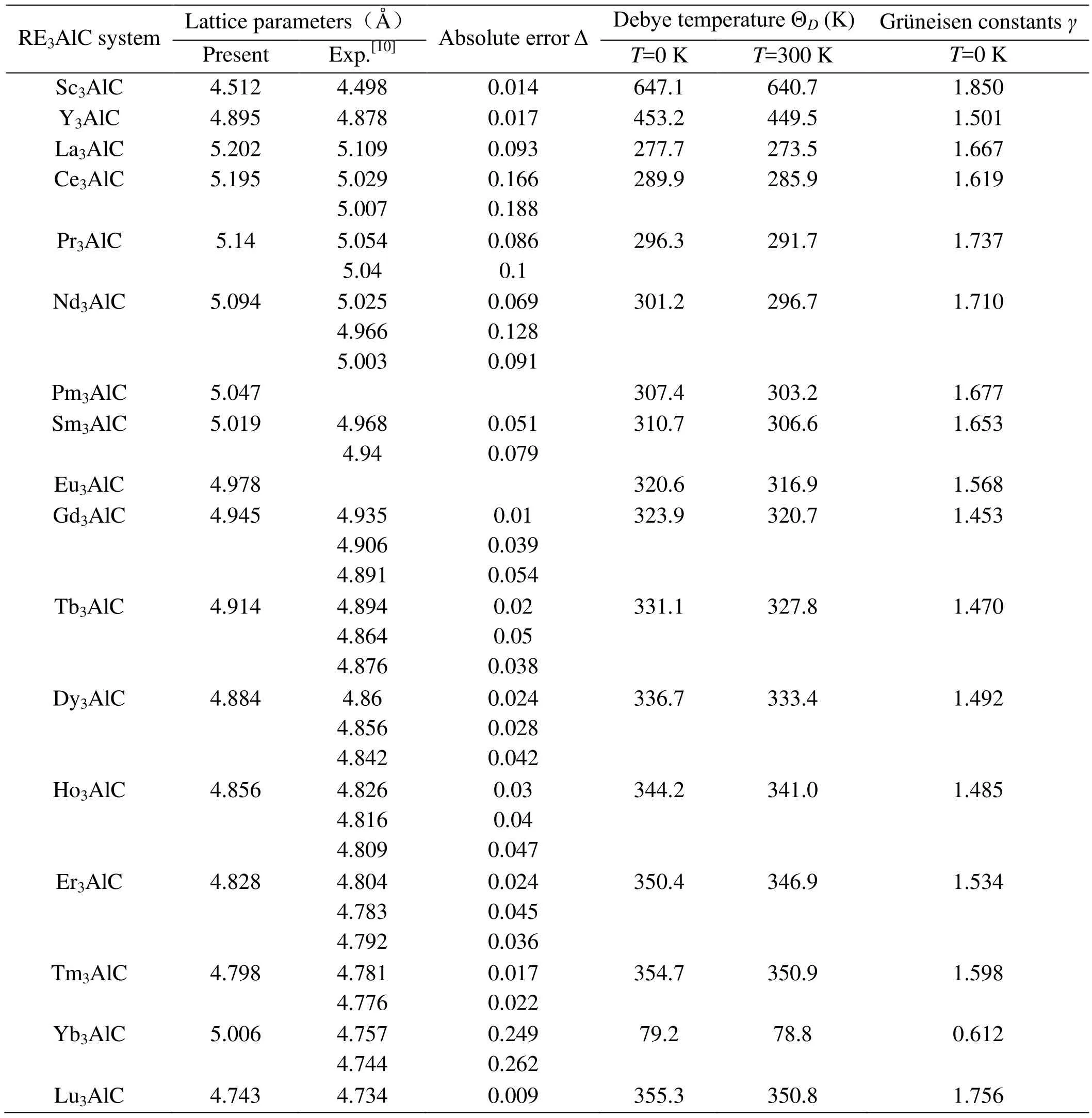
表1 RE3AlC的晶格常数、德拜温度及格律乃森常数
3.2 体积模量随温度和压强的变化
图1是体积模量随着压强和温度的变化趋势图,由图1以看出,RE3AlC金属间化合物的体积模量B随压强的增大而增大,随温度的升高而降低,符合变化规律。当T<200 K时,体积模量B随温度减小的趋势不是很明显;当T>200 K时,体积模量B随温度减小的幅度较为明显。这是因为晶格常数随温度升高而增加,晶胞体积的增加导致其体积模量减小。在一定的温度下,具有反钙钛矿结构RE3AlC化合物的体积模量B随压强的增加而增大。选取0 GPa的压强及同温度对比下,Sc3AlC的体积模量最大,Yb3AlC的体积模量最小。
3.3 吉布斯自由能随温度和压强的变化
由图2看出,在一定压强下,RE3AlC系列化合物的吉布斯自由能是随着温度的升高而降低的,并且RE3AlC系列金属间化合物各自的吉布斯自由能随温度T的变化速率大体相同,温度越高,其变化的幅度越大。在一定温度下,RE3AlC系列化合物的吉布斯自由能是随着压强的增大而升高的,其变化的趋势呈线性的增加,也就是说RE3AlC系列化合物的吉布斯自由能随压强P的变化的速率也是基本相同的。比较同温同压下的RE3AlC系列金属间化合物的各自的吉布斯自由能的大小,可知Sc3AlC化合物的吉布斯自由能是最小的,Yb3AlC化合物的吉布斯自由能是最大的,说明该系列化合物中,Sc3AlC化合物是最稳定的,Yb3AlC化合物是最不稳定的。
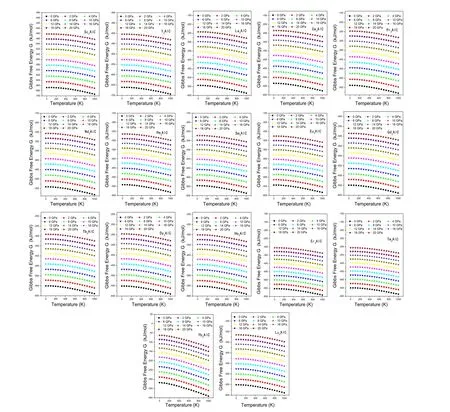
图2 吉布斯自由能随温度和压强的变化关系图
3.4 等容热容随温度和压强的变化
计算所得出的RE3AlC的等容热容的变化曲线如图3示,由图3看出,对于RE3AlC金属间化合物,其等容热容随温度和压强的变化趋势都相似,呈一个随温度的增加而增加、随压强的增加而降低的趋势。其等容热容在0-300 K温度段内变化较大,在其后的温度段内变化趋于平缓。在300 K以上的温度段,压强的变化对其比热容的影响较小,即所有的压强下其比热容趋于一个极限值。当温度很低时,其Cv值下降很快,并且T趋近于0时,Cv是正比于T的三次方,并快速接近于0。在高温度段时,其等容热容基本趋于平衡,热容值则符合杜隆-帕蒂定律,说明其在高温下的晶格比热是一个常数,与温度无关,也与物质本身所属的元素无关。

图3 定容热容和温度与压强之间的关系图
4 结论
本文结合第一性原理和准谐德拜模型计算了具有反钙钛矿结构RE3AlC(RE=Sc、Y、La、Ce、Pr、Nd、Pm、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb、Lu) 的晶格常数、德拜温度、格律乃森常数、比热容、体积模量、自由能等热物理性质。获得了体积模量、吉布斯自由能、热容量随压强和温度的变化关系。反钙钛矿结构的RE3AlC系列碳化物,其各自的比热容、体积模量、吉布斯自由能随压强和温度的变化趋势都大体相同。
[1] Blö chl P E, Fö rst C J, Schimpl J. Projector augmented wave method: ab initio molecular dynamics with full wave functions[J]. Bulletin of Materials Science, 2003, 26(1):33-41.
[2] Kresse G, Furthmü ller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Computational Materials Science, 1996, 6(1):15-50.
[3] Kresse G, Furthmü ller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54(16):11169-11186.
[4] Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review B, 1992, 45(23):13244-13249.
[5] Chadi D J. Special points for Brillouin-zone integrations[J]. Physical Review B, 1977, 16(4):5188-5192.
[6] Tao X M, Jund P, Colinet C and Tedenac J C, Phase stability and physical properties of Ta5Si3 compounds from first-principles calculations, Physical Review B, 2009, 80: 104103.
[7] Piskunov S, Heifets E, Eglitis R I, et al. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: an ab initio HF/DFT study[J]. Computational Materials Science, 2004, 29(29):165-178.
[8] Ouyang Y, Tao X, Zeng F, et al. First-principles calculations of elastic and thermo-physical properties of Al, Mg and rare earth lanthanide elements[J]. Physica B, 2009, 404(16):2299-2304.
[9] M.A. Blanco, E. Francisco,V. Luañ a. GIBBS: isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model [J]. Computer Physics Communications 158 (2004) 57-72
[10] Gesing T M, Wachtmann K H, Jeitschko W. The perovskite carbides A3MC (A = Sc, Y, La-Nd, Sm, Gd-Lu; M = Al, Ga, In, Tl, Sn, Pb)[J]. Zeitschrift fü r Naturforschung B. 1997, 52(2)
O441.3
:A
:1003-7551(2016)02-0005-06
2016-05-12
国家自然科学基金项目(11464001,51531009)及广西自然科学基金项目(2014GXNSFAA118308)
† 通讯作者:ouyangyf@gxu.edu.cn
