肉芽肿性多血管炎临床病理特点并文献复习
2017-01-16闫利娟洛阳市第二中医院病理科洛阳47003上海兰卫医学检验所病理实验室上海00335
闫利娟,朱 剑.洛阳市第二中医院病理科,洛阳47003;.上海兰卫医学检验所病理实验室,上海00335
肉芽肿性多血管炎临床病理特点并文献复习
闫利娟1,朱 剑2
1.洛阳市第二中医院病理科,洛阳471003;2.上海兰卫医学检验所病理实验室,上海200335
目的探讨肉芽肿性多血管炎的临床病理特点。方法 回顾性分析1例肉芽肿性多血管炎患者的临床、实验室检验及影像学特征并文献复习。结果肉芽肿性多血管炎患者胸部CT检查:双肺可见多结节状密度增高影,边界不清,密度不均,周围见毛刺及浅分叶,局部与胸膜相连;鼻窦CT检查左侧副鼻窦炎。实验室检查抗中性粒细胞核周抗体IgG型阳性(+)。鼻腔手术活检及肺穿刺活检均可见坏死及肉芽肿,肺活检局部可见血管炎改变。结论肉芽肿性多血管炎是一种累及呼吸道、肺和肾的坏死性肉芽肿性血管炎,发病率低,预后较差;及时明确诊断是治疗关键,在影像学及实验室检查尚不能明确诊断的情况下,可选择活检明确诊断。
肉芽肿性多血管炎;肉芽肿;血管炎;诊断
肉芽肿性多血管炎(granulomatosis with polyangiitis,GPA)是一种以坏死性肉芽肿小血管炎为特征的全身性疾病,为少见的自身免疫性疾病。临床表现复杂化、多样化,早期诊断困难较大,易被误诊或漏诊,病情进展比较迅速,如得不到及时正确的治疗,病死率高。现回顾性分析本院诊断的1例GPA病例临床病理特点,并结合相关文献进行复习。
1 临床资料
患者,女性,20岁。2015年3月因“右侧胸痛超过2个月“入院,2个月前患者睡眠时突发右侧胸痛,陈发性针刺样痛,卧位明显,深呼吸和咳嗽时加重。1个月前患者受凉后发热,测体温37.3℃,查胸部CT:左肺舌叶、右肺下叶后基底段可见结节状密度增高影,边界不清,密度不均,右肺下叶大小约5.8 cm×4.6 cm,周围见毛刺及浅分叶,局部与胸膜相连;左肺下叶见不规则软组织影,大小约1.6 cm× 1.2 cm,周围见毛刺(图1)。按“肺炎“给予抗炎治疗,胸痛稍缓解。20 d前患者再次出现咳嗽、咳少许黄痰,伴胸闷、胸痛,无发热。查抗中性粒细胞核周抗体IgG型阳性(+),抗中性粒细胞细胞质抗体IgG型阴性(-),抗髓过氧化物酶抗体IgG型1.2 U/mL,抗蛋白酶3抗体IgG型1.6 U/mL,类风湿因子(IgM亚型)31.9 U/mL,ENA抗体谱阴性(-);免疫球蛋白补体:C3(散射比浊法)1.61 g/L,C4(散射比浊法)0.39 g/L;免疫球蛋白IgA(散射比浊法)2.1 g/L,免疫球蛋白IgG(散射比浊法)12.2 g/L,免疫球蛋白IgM(散射比浊法)2.3 g/L;血常规:白细胞数10.10× 109/L,红细胞3.89×1012/L,血红蛋白111.0 g/L,血小板总数325×109/L,中性粒细胞%为79.0%,D-二聚体0.300 μg/mL,IgG4亚型:血免疫球蛋白M 1.55 g/L,血免疫球蛋白G 14.3 g/L,血免疫球蛋白A 1.68 g/L,免疫球蛋白G 40.19 g/L。在CT引导下肺穿刺病理结果显示为肉芽肿性炎,倾向自身免疫性疾病。既往3年前因感冒后开始出现双鼻塞、流涕,初鼻塞呈间歇性,交替性,涕呈粘脓性,量少,无其余不适,未引起重视,未行诊治。后鼻塞、流涕渐进性加重,鼻塞呈持续性,涕呈粘脓性,量多,频发性鼻出血,嗅觉减退。症状反复时便至当地医院对症处理。2014年12月18日因“鼻塞、多涕3年,加重1个月”入当地医院。查鼻窦CT显示,左侧副鼻窦炎,鼻中隔偏曲,双侧下鼻甲及左侧中鼻甲肥大(图2)。在全麻下行功能性鼻窦内窥镜手术。术后好转出院。术后左侧鼻腔得当地病理显示为炎性息肉,其内大量淋巴组织增生。复习鼻腔手术活检可见坏死及肉芽肿形成(图3),可见淋巴细胞、浆细胞和嗜酸性粒细胞浸润,伴血管壁多量炎性反应细胞浸润(图4)。肺穿刺活检显示,肉芽肿性炎伴坏死(图5),有多种细胞浸润的异质性炎性反应,血管壁纤维素样坏死,周围伴巨噬细胞聚集,伴有单核细胞浸润,并有上皮样细胞、多核巨细胞和成纤维细胞增生(图6),局部可见血管炎改变。免疫组化:CD20-,CD79a少量+,CD3+,CD7部分+,κ+/-,λ+/-。特殊染色:抗酸-,PAS-,六氨银-,网染+。结合病理、实验室检查及临床表现诊断肉芽肿性多血管炎,累及上呼吸道和肺。入院后多次尿常规均正常,尿红细胞位相及尿蛋白系列正常,经肾内科医师查看,考虑肾脏暂未受累。患者出院后门诊随访的一般情况较好。
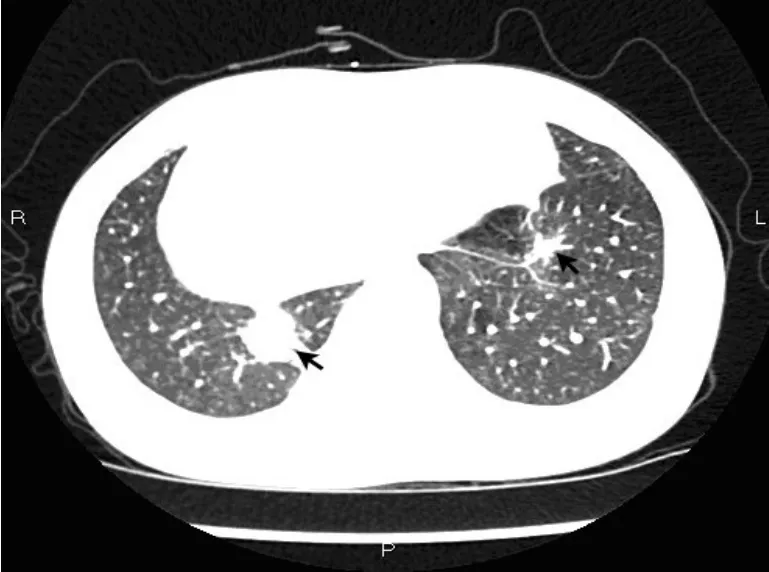
图1 CT示左、右肺下叶见不规则软组织影(箭头),局部与胸膜相连,周围见毛刺Fig.1 CT scans showed irregular soft tissue shadow under the bilateral pulmonary lobe(arrow),focal connected to the pleura,and spicula in surroundings

图2 鼻窦CT示:左侧副鼻窦炎,鼻中隔偏曲,双侧下鼻甲及左侧中鼻甲肥大(箭头)Fig.2 CT scans of paranasal sinus showed left paranasal sinusitis,deviation of nasal septum,inferior turbinate of two sides and middle turbinates hypertrophy of left side(arrow)

图3 鼻腔手术活检组织见坏死及肉芽肿形成(HE,×200)Fig.3 Necrosis and granuloma formation in nasal surgical biopsy tissues(HE,×200)

图4 高倍见肉芽肿周围见淋巴细胞、浆细胞、嗜酸性粒细胞浸润,伴血管壁多量炎症细胞浸润(HE,×400)Fig.4 High power view of granulomatosis with polyangiitis demonstrated lymph cells,plasma cells,and eosinophile granulocyte infiltration around granuloma,with inflammatory cells infiltration with the vessel wall(HE,×400)
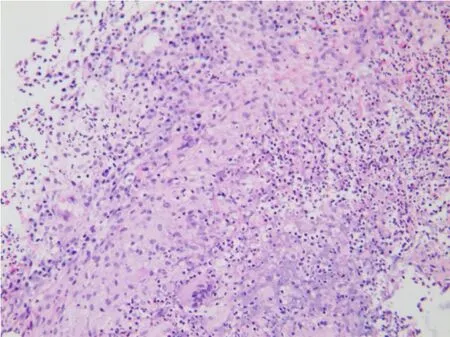
图5 肺活检标本见肉芽肿性炎伴坏死(HE,×200)Fig.5 Granulomatous inflammation with necrosis in lung biopsy tissues(HE,×200)
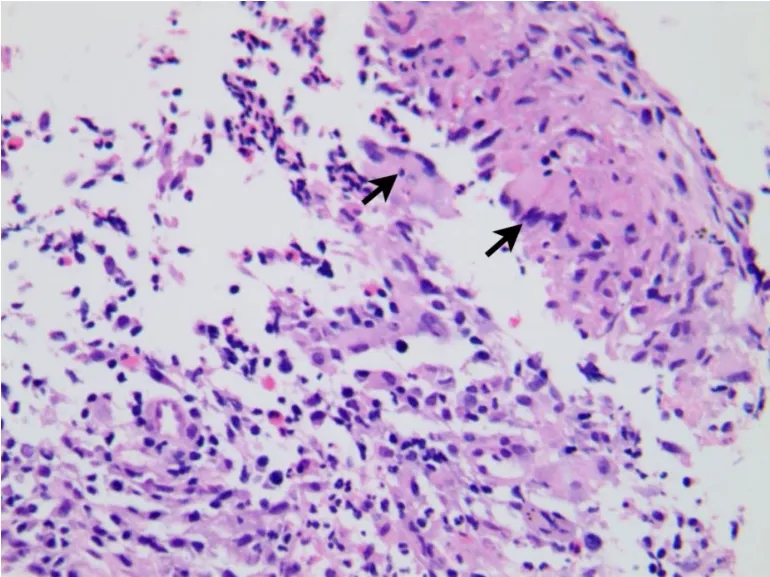
图6 有多种细胞浸润的异质性炎性反应,血管壁纤维素样坏死,周围伴巨噬细胞聚集,伴有单核细胞浸润,并有上皮样细胞、多核巨细胞(箭头)及成纤维细胞增生(HE,×400)Fig.6 Heterogeneous inflammatory reaction with a variety of cell infiltration,fibrinoid necrosis surrounded by macrophages,mononuclear cell infiltration,with epithelioid cells,multinucleated giant cells(arrow)and fibroblasts hyperplasia(HE,×400)
2 讨论
GPA旧称韦 格 纳肉芽肿病(Wegener granulomatosis,WG),由德国医师Friedrich[1]在1936年和1939年详细阐述了该病的病理特征而得名,2009年,Gómez-Puerta等[2]在CHEST发表专文,提出 用 坏 死 性 肉 芽 肿 性血 管 炎(necrotizing granulomatous vasculitis,NGV)取代WG,但未获得广泛采纳。2011年初,美国风湿病学会、美国肾脏病学会及欧洲风湿病学会联名提出将“WG”这一以人名命名的疾病名称更新为“GPA”[3]。更名最主要原因是近20年来人们对本病病因、发病机制及病理学特点认识的提高。这一新的命名逐渐被广泛接受。
GPA临床上男性稍多于女性,年龄5~91岁,40~50岁是疾病高发年龄。临床表现主要为三联征,即上呼吸道、下呼吸道和肾脏损伤。上呼吸道病变主要为鼻窦炎、鼻腔病变、中耳炎、耳痛、听力下降、声门狭窄、咳嗽和口腔病变等,超过80%的GPA患者会累及鼻腔鼻窦[4]。下呼吸道病变表现为咳嗽、咯血、胸痛和发热,75%的GPA患者出现肺影像学异常,可出现咯血、贫血和呼吸困难,即GPA所致的弥漫性肺泡出血表现,部分患者可出现胸腔积液,常单侧。肾脏损伤表现为血尿、蛋白尿和红细胞型尿。其他可见关节痛、外周和中枢神经病变、心包炎及皮肤有红斑、瘀斑和结节等。GPA诊断难度较大,若对本病缺乏认识,就很容易误诊漏诊,本例患者是以上呼吸道鼻腔鼻窦病变为首发,出现下呼吸道病变后才予以明确诊断。
影像学上鼻部早期鼻窦CT表现与普通鼻窦炎类似,较难鉴别,可见鼻黏膜增厚、副鼻窦软组织影、积液及息肉等;中晚期可见中线结构破坏、鼻窦骨质破坏、异常骨新生、骨性鼻中隔及鼻甲破坏消失等。肺部X线胸片显示胸部浸润影和结节影。CT显示两侧肺内多发性、多形性和多变性的结节或肿块影,可有空洞形成,即所谓“三多一洞”,也可见毛玻璃影、肺不张或支气管束增宽支气管狭窄等[5]。影像学可帮助判断疾病的活动性。毛玻璃影和>3 cm的结节影同时存在预示疾病的活动期。本病例的鼻部及肺部影像学比较相符。
实验室检查抗中性粒细胞胞质抗体(antineutrophil cytoplasmic antibodies,ANCA)可呈阳性。ANCA是一组与中性粒细胞或单核细胞细胞质中的一些特异性抗原发生反应的自身抗体,在荧光显微镜下,根据荧光分布主要分为胞质型ANCA(c-ANCA)、核周型ANCA(p-ANCA)和非典型ANCA(a-ANCA或x-ANCA)。其靶抗原包括多种物质,如蛋白酶-3(PR-3)、髓过氧化物酶(MPO)、弹性蛋白酶、乳铁蛋白和组织蛋白酶等。c-ANCA靶抗原主要为PR-3。ANCA检测对诊断GPA具有快速、简单、特异度和灵敏度高的优势,尤其是c-ANCA阳性,诊断GPA特异度达95%~98%[6]。c-ANCA阳性与呼吸道和眼部受累密切相关,而且与病情活动性相关。其余实验室检查异常包括白细胞升高、红细胞沉降率增快、C反应蛋白升高和肾功能不全等。本例患者抗中性粒细胞核周抗体IgG型阳性(+),其余实验室检查未见明显异常。
典型的GPA主要病理特征为肉芽肿、血管炎和局灶性坏死三联征。主要病变部位包括小动脉、静脉及毛细血管,偶亦可累及大动脉。GPA肉芽肿特征与传统意义上的肉芽肿是不同的,前者肉芽肿是有多种细胞浸润的异质性炎性反应,其中心常存在血管壁纤维素样坏死,周围以巨噬细胞聚集为主,伴有单核细胞浸润,并有上皮样细胞、多核巨细胞及成纤维细胞增生,细胞聚集较多时,即被称为肉芽肿性结节;后者肉芽肿主要是由巨噬细胞及其衍生细胞局限性浸润和增生所形成的边界清楚的结节状病灶。血管炎可以发生在动脉、静脉和毛细血管上,直径多<0.5 cm。病变中常不止一种类型血管受累,主要是血管慢性炎性反应,指血管壁全层有淋巴细胞和浆细胞为主的炎细胞浸润,弹力纤维染色可见血管壁弹力纤维破坏;也可以是急性炎性反应,指血管壁全层有以中性粒细胞为主的炎细胞浸润;血管炎类型包括:①坏死性纤维素性血管炎指血管壁有坏死伴有红染的纤维素渗出;②肉芽肿性血管炎指血管壁可见以多核巨细胞为标志的组织细胞浸润或是出现栅栏状组织细胞围绕;③瘢痕性血管炎指血管壁肌层或内膜增厚纤维化,管腔狭窄,少量慢性炎细胞浸润,可能是一种治疗改变。坏死可以形成中性粒细胞微脓肿和地图样坏死,中性粒细胞微脓肿可能是坏死的早期形式,直径≤1 mm,呈针状或斑点状;地图样坏死中央为嗜碱性、颗粒状,伴有不规则边界,形状不规则,直径>1 mm,坏死结节周围是栅栏状排列的组织细胞和多核巨细胞。地图样坏死是传统上强调的坏死类型,当缺乏时,出现中性粒细胞微脓肿也需要考虑GPA诊断。除典型的病理特征外,亦可出现间质纤维组织增生、淋巴细胞聚集及嗜酸性粒细胞浸润,病变累及肺脏可导致机化性肺炎、肺泡出血和弥漫性肺出血,少见的有脂质性肺炎、急慢性支气管炎,内源性脂质性肺炎和阻塞性细支气管炎等病理改变。GPA的病理改变复杂,诊断困难,除了其本身有复杂的形态学表现外,当以某种组织学改变为主时,需要与相关的病变相鉴别:①感染性肉芽肿,诊断GPA前必须排除感染,很多感染均可形成坏死和肉芽肿性炎性反应,在坏死灶内或周围常有血管炎[7],最需鉴别的结核肉芽肿常见于肺上叶尖端及背端,此时只能进行特殊染色病原体培养和结合临床生化检查进行鉴别;②变应性血管炎性肉芽肿(Churg-Strauss综合征:病理特点为大量的嗜酸性粒细胞渗出、血管外的肉芽肿性炎性反应和中小血管的坏死性炎性反应,在肺部主要与哮喘和嗜酸性粒细胞增多相关,在GPA组织中常有多少不等的嗜酸性粒细胞浸润,当伴有明显嗜酸性粒细胞浸润时应和Churg-Strauss综合征鉴别,血清学检查大约50% Churg-Strauss综合征病例ANCA阳性,主要为MPO-pANCA阳性,而GPA侧为c-ANCA阳性;③支气管中心性肉芽肿病:病理表现的突出特征为富含嗜酸性粒细胞的非干酪性肉芽肿,肉芽肿周围可见有血管炎的表现,与GPA血管炎不同的是无血管中心破坏,出现坏死可见真菌菌丝,无血ANCA升高;④淋巴瘤样肉芽肿病:这是最常累及肺的EB病毒相关的淋巴组织增生性病变,明显的组织改变是血管中心性淋巴组织浸润,当伴有组织坏死时易与GPA混淆,淋巴瘤样肉芽肿缺乏肉芽肿性炎性反应,无血ANCA升高;⑤显微镜下多血管炎:这是一种非肉芽肿性ANCA相关小血管炎,其胸片示双侧片状或弥散性实变,CT显示磨玻璃影、实变影和支气管血管增厚,无结节影,影像学特点主要与肺泡出血、间质炎性反应和纤维化有关,实验室检查MPO-pANCA阳性。
GPA如不经及时治疗5年生存率只有10%,平均存活时间为5个月,死因通常是肾衰竭和/或呼吸衰竭。目前主要治疗采用糖皮质激素联合免疫抑制剂(如环磷酰胺和甲氨蝶呤)联合治疗,疗程不少于18个月[8],有效减少病死率、延长缓解期,5年生存期提高到80%以上[9]。对于难治性、复发性GPA,采用B细胞抗体利妥昔单抗能够缓解[10]。预后不仅取决于病变本身,还包括治疗对其他器官的损害,包括肺功能不全、肾功能不全、糖皮质激素性糖尿病和骨质疏松等。因此,密切随访对提高治疗效果至关重要。GPA涉及多个学科,提高诊断意识,尽早诊断和及时合理治疗是改善预后、延长生存期的关键。
[1] Friedrich.Illustrated histopathologic classification criteria for selected vasculitis syndromes[J].Arthritis Rheum,1990,33(8):1074-1087.
[2] Gómez-Puerta JA,Hernández-Rodríguez J,López Soto A,et al. Antineutrophilcytoplasmic antibody-associated vasculitides and respiratory disease[J].Chest,2009,136(4):1101-1111.
[3] Falk RJ,Gross WL,Guillevin L,et al.Granulomatosis with polyangiitis(Wegener's):an alternative name for Wegener's granulomatosis[J].Arthritis Rheum,2011,63(4):863-864.
[4] Janisiewicz AM,Klau MH,Keschner DB,et al.Higher antineutrophilcytoplasmicantibody(C-ANCA) titersare associated with increased overall healthcare use in patients with sinonasal manifestations of granulomatosis with polyangiitis(GPA)[J].Am J Rhinol Allergy,2015,29(3):202-206.
[5] Semenkova EN,Krivosheev OG,Novikov PI,et al.Pulmonary lesions in Wegener's granulomatosis[J].Klin Med(Mosk),2011,89(1):10.
[6] Lamprecht P,GrossWL.Wegener's Granulomatosis[J].Herz,2004,29(1):47-56.
[7] Martinez F,Chung JH,Digumarthy SR,et al.Common and uncommon manifestations of Wegener granulomatosis at chest CT:radiologic-pathologic correlation[J].Radiographics,2012,32(1):51-69.
[8] 蔡柏蔷,李龙芸.呼吸病学[M].2版.北京:协和医科大学出版社,2010:1558-1559.
[9] Jayne D.Part3:Newer therapies for ANCA-associated vasculitis [J].Clin Exp Rheumatol,2007,25(1 Suppl 44):S77-S79.
[10] Fervenza FC.Rituximab in ANCA-associated vasculitis:fad or fact[J].Nephron Clin Pract,2011,118(2):c182-c188.
Granulomatosis with polyangiitis:clinicopathological features and literature review
YAN Lijuan1,ZHU Jian2
1.Department of Pathology,Luoyang the Second Hospital of Traditional Chinese Medicine,Luoyang 471003,China;2.Laboratory of Pathology,Labway of Shanghai,Shanghai 200335,China
ObjectiveTo explore the clinicopathological features of granulomatosis with polyangiitis.MethodsThe clinical manifestations,laboratory findings and radiological features of one case of granulomatosis with polyangiitis were retrospectively analyzed,and the related literatures were reviewed.ResultsComputed tomography of the chest showed bilateral pulmonary multinodular high density shape,with obscure margin and inhomogeneous density,and spicula and lobe were seen in surroundings,with part adhering to the pleural.Sinuses computed tomography demonstrated left paranasal sinusitis.The laboratory examination revealed a positive result of anti-neutrophil cytoplasmic antibody IgG.Biopsy samples were obtained from nasal cavity operation and lung puncture,and necrotizing granuloma and local changes in vasculitis were observed.ConclusionGranulomatosis with polyangiitis is a kind of necrotizing granulomatous vasculitis,which involves respiratory tract,lung and kidney with low incidence and poor prognosis. Biopsy can be conducted to confirm diagnosis in case of failed definitive diagnosis by laboratory and radiological examinations.
Granulomatosis with polyangiitis;Granuloma;Vasculitis;Diagnosis
R593.2
A
2095-378X(2016)01-0044-05
10.3969/j.issn.2095-378X.2016.01.015
2015-11-12)
闫利娟(1972—),女,主治医师,从事临床病理诊断工作
朱 剑,电子信箱:zhujian770@aliyun.com
