头孢地尼在健康人体中生理药动学模型的建立与应用Δ
2017-01-06汪难喜翟学佳朱超然张新林吕永宁华中科技大学同济医学院附属协和医院药剂科武汉430022
汪难喜,翟学佳,朱超然,张新林,吕永宁(华中科技大学同济医学院附属协和医院药剂科,武汉 430022)
·临床药学与研究·
头孢地尼在健康人体中生理药动学模型的建立与应用Δ
汪难喜*,翟学佳,朱超然,张新林,吕永宁#(华中科技大学同济医学院附属协和医院药剂科,武汉 430022)
目的:建立头孢地尼在健康人体内的生理药动学(PBPK)模型,预测其口服后在人体内的药动学过程。方法:以“头孢地尼”“cefdinir”“logP”“pKa”等为关键词,检索中国知网、ScienceDirect和PubMed等数据库关于头孢地尼理化常数的相关文献,遵照美国食品与药物管理局的相关指导原则和前期临床试验方案,采用GastroPlusTM8.6软件建立头孢地尼口服给药的PBPK模型,并通过倍数误差来评价模型的有效性;运用该PBPK模型模拟头孢地尼在胃肠道吸收情况;以头孢地尼参比制剂(颗粒剂和胶囊剂)释放速率t85%=15 min(即累积溶出85%的时间为15 min)时的cmax和AUC0-∞为参比,通过单次和群体(n=500)模拟试验,评价受试制剂与参比制剂的生物等效性。结果:PBPK模型预测头孢地尼的药-时曲线与实测值拟合良好,R2≥0.95;药动学参数cmax、tmax、AUC0-∞与实测值接近,倍数误差<2。口服给药后,头孢地尼主要在肠道吸收(45.6%),主要吸收部位为空肠1段(14.8%);其吸收量明显低于吸收部位的释放量,4 h左右达到最大吸收分数(约40%)。单次模拟试验结果显示,受试制剂与参比制剂的cmax和AUC0-∞比较,差异均无统计学意义(P>0.05);群体模拟试验结果显示,受试颗粒和受试胶囊的相对生物利用度分别为99.01%~102.99%、97.60%~105.90%,其cmax和AUC0-∞的90%置信区间均在参比制剂相应参数的80%~125%之内。结论:该PBPK模型准确可靠,可为头孢地尼制剂的药动学研究和生物等效性评价提供参考。受试制剂与参比制剂生物等效。
生理药动学模型;头孢地尼;药动学;胃肠吸收;生物等效性
头孢地尼抗菌谱广、不良反应少,在临床上应用广泛[1]。国内已有多家制药公司对其进行仿制,剂型主要包括颗粒剂、胶囊剂等,质量不一。目前,已有关于其制剂质量控制的文献报道,但仅通过体外溶出试验评价其质量[2]。然而,由于头孢地尼在生物药剂学分类系统(BCS)中属Ⅳ类药物,具有低溶解性、低渗透性的特点,体内吸收是关键的限速因素。因此,仅以体外溶出试验预测其制剂质量一致性的能力十分有限[3]。故本研究在此基础上,首次建立了餐后单次口服头孢地尼颗粒和胶囊在健康人体内的生理药动学(PBPK)模型,并通过前期临床试验所得的药动学结果评价其准确性,为头孢地尼仿制制剂生物等效性的初步评价与上市药品质量再评价提供参考。
1 材料
GastroPlusTM8.6软件、ADMET PredictorTM7.1软件(美国SimulationPlus公司);ZRS-4型智能溶出试验仪(天津大学精密仪器厂);UV2450型紫外可见光谱仪、AUW220D型电子分析天平(日本岛津公司);PHS-3B型精密pH计(上海精科雷磁公司)。
受试制剂:头孢地尼颗粒(批号:311130108,规格:50 mg/袋)、头孢地尼胶囊(批号:310130107,规格:100 mg/粒)均由石家庄市华新药业有限责任公司提供;参比制剂(即原研制剂):头孢地尼颗粒(批号:025310,规格:100 mg/袋)、头孢地尼胶囊(批号:023730,规格:100 mg/粒)均购自日本藤泽公司;头孢地尼对照品(中国食品药品检定研究院,批号:130502-201403,纯度:98%);磷酸、甲醇、乙腈为色谱纯,盐酸、氢氧化钠、磷酸二氢钾为分析纯,水为超纯水。
2 方法
2.1 PBPK模型参数
以“头孢地尼”“cefdinir”“logP”“pKa”等为关键词,检索2005-2015年中国知网(CNKI)、ScienceDirect和PubMed等数据库中的相关文献资料,得其理化常数如下:相对分子质量为395.41,溶解度为0.988 mg/ml,脂水分布系数(logP)为-0.16,各级酸碱解离常数(pKa)为1.9/3.3/9.9[4-5]。平均沉降时间、颗粒密度、人表观吸收系统值(Peffa)均由ADMET PredictorTM7.1软件进行预测,分别为0.75×105cm2/s、1.2 g/ml、0.25×2-4cm2/s。
遵照美国食品与药物管理局(FDA)的相关指导原则[6]及本课题组前期临床试验方案[7-8],将受试者给药剂量设定为100 mg,送服药物时的饮水体积设定为250 ml,受试者年龄设定为(22.13±1.48)岁,体质量设定为(65.04±6.52)kg,服药方式设定为餐后单次口服(受试者资料以及临床试验方案详见本课题组前期已发表的头孢地尼人体药动学研究文献[7-8])。
2.2 PBPK模型的建立
将“2.1”项下头孢地尼的各项理化参数和临床给药方案输入至GastroPlusTM8.6软件内,并在软件的“剂型选项”中分别选择颗粒剂与胶囊剂。同时,根据临床试验中健康受试者的资料选择相应的生理学与解剖学参数库,从而获得各组织器官的体积、血流灌注速率和组织血浆分布系数(Kp)。本模型共包含由肝脏、肾脏组成的2个消除器官室和12个非消除器官室等14个房室,各房室皆采用动静脉血液循环相连接,此时任一时刻各组织器官室中的药物浓度均是动态平衡的[9]。由于头孢地尼分布到各组织器官室的速率主要受透膜的影响[3],因此在进行PBPK模型分布参数的设置时,选择“透膜限速”项。试验建模过程中,各项软件皆于Windows 7系统环境下运行。
2.3 PBPK模型的有效性评价
采用GastroPlusTM8.6软件中的“卷积分项”,通过加载头孢地尼的理化参数和人体胃肠道生理参数,建立了餐后健康人体中头孢地尼口服给药的PBPK模型。通过模型模拟了餐后分别口服头孢地尼2种制剂在人体内的药-时曲线;在软件中输入本课题组前期临床试验所得的药动学数据[7-8],进行曲线拟合度评价,以倍数误差来评价各参数的拟合度。若模型预测的cmax和AUC0-∞在实测值的2倍以内,则认为拟合度良好,表明该预测模型可靠、有效[9]。
2.4 药物释放与胃肠道吸收模拟
通过头孢地尼PBPK模型中的“基础模块”,运用模拟口服制剂胃肠道吸收的高级房室模型(ACAT),预测头孢地尼在胃、十二指肠、空肠等部位的溶出和吸收情况,计算出头孢地尼在胃肠道各部位的吸收量、释放量和吸收分数,并通过“卷积分项”绘制吸收与释放曲线。
2.5 应用PBPK模型评价受试制剂与参比制剂的生物等效性
应用PBPK模型模拟了释放速率对制剂生物等效性的影响,初步探究满足生物等效性标准时头孢地尼口服制剂所能接受的最低释放速率。参照2015年版《中国药典》[10],采用浆法测定受试制剂与参比制剂的体外溶出度:随机选取受试制剂与参比制剂各6颗(袋),分别置于4种不同介质(介质1~4分别为水、pH值为6.8的KH2PO4溶液、pH值为4.5的KH2PO4溶液、pH值为1.0的HCl溶液)中,转速为50 r/min,温度为(37.0±0.5)℃。分别于5、10、15、20、30 min吸取溶出液各5 ml(同时补液5 ml),采用标准曲线法测定头孢地尼的溶出量。将实测的体外溶出数据加载至PBPK模型中,进行单次和群体(n=500)模拟试验,考察受试制剂与参比制剂的体内药动学特征,并评价其生物等效性。以头孢地尼参比制剂(颗粒剂和胶囊剂)释放速率为t85%=15 min(即累积溶出85%的时间为15 min)时的cmax和AUC0-∞作为参比,比较具有不同释放速率的头孢地尼口服制剂的cmax和AUC0-∞,若其cmax和AUC0-∞的90%置信区间(CI)均在参比制剂相应参数的80%~125%范围内,则认为受试制剂与参比制剂生物等效[11]。
2.6 统计学分析
3 结果
3.1 PBPK模型的有效性评价结果
结果显示,运用PBPK模型预测餐后单次口服头孢地尼的药-时曲线与实测药-时曲线拟合良好(见图1),R2≥0.95。

图1 PBPK模型预测头孢地尼制剂的药-时曲线与实测药-时曲线比较A.颗粒剂;B胶囊剂Fig 1 Comparison of blood concentration-time curve predicted by PBPK model and measured blood concentration-time curveA.particle;B.capsule
模拟结果显示,模型预测头孢地尼胶囊剂和颗粒剂的cmax、tmax和AUC0-∞均与实测值[7-8]接近,倍数误差<2(见表1),表明该模型准确性良好,可用于模拟不同头孢地尼制剂在人体内的药动学行为。

表1 PBPK模型预测头孢地尼的药动学参数与实测值比较Tab 1 Comparison of pharmacokinetic parameters of cefdinir predicted by PBPK model and measured value
3.2 PBPK模型模拟头孢地尼口服后的吸收与释放
模拟结果显示,口服给药后,头孢地尼主要在肠道吸收(45.6%),主要的吸收部位为空肠1段(14.8%),提示对于空肠病变患者,其头孢地尼的吸收量减少,可能会导致治疗失败。此外,口服头孢地尼制剂后,其吸收量远远低于吸收部位的释放量,4 h左右达到最大吸收分数,约为40%,与文献[12]基本一致。PBPK模型预测头孢地尼口服后的吸收情况见图2;PBPK模型预测头孢地尼的吸收与释放曲线见图3。

图2 PBPK模型预测头孢地尼口服后的吸收情况Fig 2 The absorption of cefdinir after administration predicted by PBPK model

图3 PBPK模型预测头孢地尼的吸收与释放曲线Fig 3 The absorption-desorption curve predicted by PBPK model
3.3 PBPK模型研究药物体内释放速率对其生物等效性的影响
模拟试验中,将人体生理参数和药物理化参数等均设定在其预设值的10%误差范围内随机波动。结果显示,当头孢地尼口服制剂的释放速率为t85%=90 min时,无论是cmax还是
AUC0-∞,均在参比制剂相应参数的80%~125%范围内,两者生物等效(见图4)。同时,根据世界卫生组织(WHO)的生物豁免原则,若药物释放快速(t85%≤15 min),则认为其在体内的吸收特征类似于口服溶液制剂,则可免除生物等效性试验[13]。
本研究结果表明,只有当头孢地尼口服制剂在体内的释放速率不低于t85%=90 min,才可获得生物豁免。提示体外溶出速率可能对其生物等效性产生影响,t85%=90 min可作为最低释放速率成为初步评价制剂质量的参考之一。
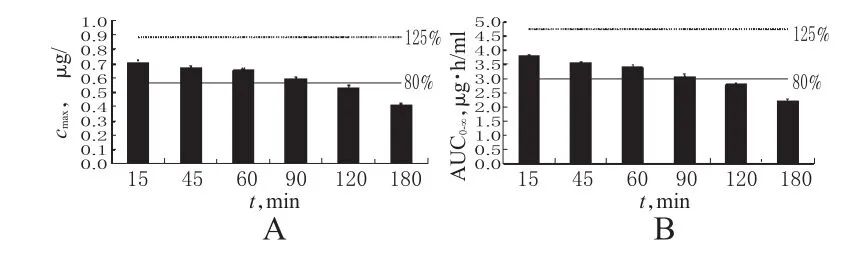
图4 头孢地尼在不同释放速率下的cmax和AUC0--∞A.cmax;B.AUC0-∞注:实、虚线分别表示在t85%=15 min时,头孢地尼参比制剂cmax和AUC0-∞的80%和125%,在此范围内则表明受试制剂与参比制剂生物等效Fig 4 cmaxandAUC0--∞of cefdinir at different release ratesA.cmax;B.AUC0-∞Note:solid and broken lines respectively represent 80%and 125%of cmaxand AUC0-∞of cefdinir reference preparations when t85%=15 min.In this range means test preparation and reference preparation are bioequivalent
3.4 PBPK模型预测不同头孢地尼制剂在健康人体内的生物等效性
体外溶出试验显示,30 min内受试制剂与参比制剂的累积溶出度分别为89.92%~103.80%、89.53%~102.06%。当溶出时间为5 min时,受试胶囊与参比胶囊在介质2中的累积溶出度比较,差异有统计学意义[(73.54±3.23)%vs.(8.77± 1.11)%,P<0.05],表现为受试胶囊的溶出速率较参比胶囊快;当溶出时间为15 min时,受试制剂与参比制剂在4种介质中的累积溶出度均超过85%,差异无统计学意义(P>0.05)。
受试制剂与参比制剂的体外溶出虽基本相似,但仍表现出一定的差异。将各制剂在不同介质中的实测溶出数据加载至PBPK模型中,进行单次和群体模拟试验,进一步考察受试制剂与参比制剂的生物等效性。
单次模拟试验结果显示,受试颗粒/胶囊和参比颗粒/胶囊体内生物利用度的模拟结果均>42%,基本达到文献记载的最大值[12]。单次模拟试验模拟所得的受试制剂cmax和AUC0-∞与参比制剂比较,差异均无统计学差异(P>0.05),提示两者可能具有等效性。单次模拟试验模拟所得头孢地尼制剂的cmax和AUC0-∞见表2。

表2 单次模拟试验模拟所得头孢地尼制剂的cmax和AUC0--∞Tab 2 cmaxand AUC0--∞of cefdinir preparation simulated in single simulation test
群体模拟试验结果显示,受试颗粒/胶囊在4种介质中的相对生物利用度分别在99.01%~102.99%、97.60%~105.90%。将对数转换后受试制剂和参比制剂的cmax和AUC0-∞进行方差分析,采用双向单侧t检验及[1-2α]%CI法进行生物等效性评价。结果显示,受试制剂的cmax和AUC0-∞的90%CI均在参比制剂相应参数的80%~125%范围内,均拒绝生物不等效假设,提示受试制剂与参比制剂生物等效,详见表3。
4 讨论
本研究根据头孢地尼的理化性质和受试者生理、生化数据,采用GastroPlusTM8.6软件成功建立了其在健康人体内的PBPK模型,并通过倍数误差来评价模型的有效性。结果显示,该PBPK模型的预测值与前期临床试验中健康人餐后单次口服头孢地尼的实测值十分接近,两者拟合良好。在模拟餐后头孢地尼胶囊的药动学特征时,虽然其tmax实测值与预测值之间存在一定差异(倍数误差为1.27),但误差仍在实测值的2倍以内,可认为两者拟合良好。造成实测tmax较预测值相对延后(见图1A、表1)的原因可能与其血浆蛋白结合有关。头孢地尼经胃肠道吸收后,约有70%的药物会与血浆蛋白结合,且不同剂量、不同剂型均可一定程度地影响血浆蛋白结合速率及其程度,进而造成tmax延后[12,14]。

表3 群体模拟试验模拟所得头孢地尼的cmax和AUC0--∞及生物等效性评价Tab 3 cmax,AUC0--∞of cefdinir preparation simulated in population simulation test and bioequivalence evaluation
运用该PBPK模型模拟头孢地尼在胃肠道的吸收与释放,发现该药主要在肠道吸收(45.6%),主要吸收部位为空肠1段,提示对于空肠病变患者,临床应注意用药方案的调整。同时,头孢地尼作为低渗透药物,服药后4 h左右吸收达到最大值,且口服吸收量远远低于其吸收部位的释放量,可通过制剂手段降低药物的释放速率,以增加其吸收程度。
PBPK模型借助药物体外溶出数据,特异性模拟其在体内的药动学行为,现已被广泛应用于药物体内外相关性(IVIVC)分析和生物等效性评价[15]。本研究运用PBPK模型,结合头孢地尼制剂的体外溶出试验,初步评价了头孢地尼受试制剂与参比制剂的生物等效性。本文参照日本橙皮书选择相应的介质进行体外溶出试验,结果表明两种制剂的体外溶出行为在介质2中的前5 min内存在一定差异,提示该介质可能是最能反映头孢地尼制剂体内释放特性的溶出介质。单次和群体模拟试验结果显示,受试制剂与参比制剂的cmax和AUC0-∞比较,差异均无统计学意义(P>0.05);受试颗粒和受试胶囊的相对生物利用度分别为99.01%~102.99%、97.60%~105.90%,其cmax和AUC0-∞的90%CI均在参比制剂相应参数的80%~125%之内,两者生物等效。
PBPK模型基于真实生理或解剖学特征,建立预测模型模拟药物在体内的代谢过程,研究者可根据实际情况调整模型中的相应参数,故利用该模型可预测头孢地尼在不同生理及病理条件、药物剂型、给药剂量、给药途径等情况下药动学参数的变化,进而指导临床合理用药[16]。由于头孢地尼的理化参数均来源于文献,因此模拟结果尚存在一定的局限性,在后续研究中,应进一步通过试验补充相应数据,使头孢地尼制剂在健康人体内的PBPK模型趋于完善。
[1] Sader HS,Jones RN.Cefdinir:an oral cephalosporin for the treatment of respiratory tract infections and skin and skin structrue infections[J].Expert Rev Anti Infect Ther,2007,5(1):29.
[2] 赵洪霞,姜起栋,梁丽娟,等.不同介质中头孢地尼干混悬剂溶出曲线相似性比较的研究[J].天津药学,2014,26(4):16.
[3] 付晓峰,柯学.口服药物溶出技术与体内外相关性的研究进展[J].药学与临床研究,2012,20(2):142.
[4] National Center for Biotechnology Information.Cefdinir [EB/OL].[2015-11-03].http://pubchem.ncbi.nlm.nih.gov/ compound/6915944#section=Top.
[5] DrugBank.Cefdinir[EB/OL].[2015-11-03].http://www. drugbank.ca/drugs/DB00535#pharmacology.
[6] U.S.Department of Health and Human Services,Food and Drug Administration,Center for Evaluation and Research.Guidance for industry:waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System[S].2000-08-02.
[7] 陈芬,朱超然,翟学佳,等.头孢地尼胶囊在健康人体的生物等效性研究[J].医药导报,2015,34(10):1 288.
[8] 汪难喜,翟学佳,朱超然,等.液相色谱-串联质谱联用法测定人血浆中头孢地尼的浓度及生物等效性研究[J].中国医院用药评价与分析,2016,16(8):1 014.
[9] Yamazaki S,Skaptason J,Romero D,et al.Prediction of oral pharmacokinetics of cMet kinase inhibitors in humans:physiologically based pharmacokinetic model versus traditional one-compartment model[J].Drug Metab Dispos,2011,39(3):383.
[10] 国家药典委员会.中华人民共和国药典:二部[S].2015年版.北京:中国医药科技出版社,2015:177.
[11] Guay DR.Pharmacodynamics and pharmacokinetics of cefdinir,an oral extended spectrum cephalosporin[J].Pediatr Infect Dis J,2000,19(12):S141.
[12] Tsume Y,Amidon GL.The biowaiver extension for BCS classⅢdrugs:the effect of dissolution rate on the bioequivalence of BCS classⅢimmediate-release drugs predicted by computer simulation[J].Mol Pharm,2010,7(4):1 235.
[13] World Health Organization.Proposal to waive in vivo bioequivalence requirements for WHO Model List of Essential Medicines immediate-release,solid oral dosage forms [S].2006-08-30.
[14] 郭宾,李川.药物与血浆蛋白结合的药理学基础及其研究进展[J].中国临床药理学与治疗学,2005,10(3):241.
[15] Kovacević I,Parojcić J,Homsek I,et al.Justification of biowaiver for carbamazepine,alow soluble high permeable compound,in solid dosage formsbased on IVIVC and gastrointestinal simulation[J].Mol Pharm,2009,6(1):40.
[16] Rowlabd M,Peck C,Tucker G.Physiologically-based pharmacokinetics in drug development and regulatory science[J].Annu Rev Pharmacol Toxicol,2011,doi:10.1146/ annurev-pharmtox-010510-100540.
Establishment of Physiological Pharmacokinetic Model of Cefdinir in Healthy Volunteers and ItsApplication
WANG Nanxi,ZHAI Xuejia,ZHU Chaoran,ZHANG Xinlin,LYU Yongning(Dept.of Pharmacy,Union Hospital,Tongji Medical College,Huazhong University of Science and Technology,Wuhan 430022,China)
OBJECTIVE:To establish physiological pharmacokinetic(PBPK)model of cefdinir in healthy volunteers,and to predict pharmacokinetic process of cefdinir in volunteers after oral administration.METHODS:Using“toubao dini”“cefdinir”“logP”“pKa”as keywords,related literatures about physico-chemical constants of cefdinir were retrieved from CNKI,ScienceDirect,PubMed and other databases;according to related guidelines and preliminary clinical trial plan of FDA,GastroPlusTM8.6 software was used to establish PBPK model of oral administration of cefdinir;the effectiveness of the model was evaluated by multiple error.The model was used to simulate the absorption of cefdinir in the gastrointestinal tracts.The bioequivalence of test preparation and reference preparation were evaluated through single and population(n=500)simulation tests using cmaxand AUC0-∞of cefdinir reference preparation(capsule and granular formulation)as factors when release rate t85%=15 min(i.e.accumulatively released 85%within 15 min).RESULTS:The blood concentration-time curves of cefdinir predicted by PBPK model fitted well with measured value(R2≥0.95);the pharmacokinetic parameters(cmax,tmax,AUC0-∞)were close to measured results,and the multiple errors were less than 2.After oral administration,cefdinir was mainly absorbed by the intestinal tract(45.6%),especially by segment 1 of jejunum(14.8%);the absorption amount was significantly lower than the release amount of absorption site,and reached the maximal value(about 40%)within 4 h.The results of single simulation test showed that there was no statistical significance in cmaxand AUC0-∞between cefdinir test and reference preparations(P>0.05).The results of population simulation test showed that the relative bioavailability of cefdinir test particle and test capsule respectively were 99.01%-102.99%and 97.60%-105.90%;90%CI of cmaxand AUC0-∞values were within 80%-125%of reference preparation.CONCLUSIONS:The PBPK model is accurate and reliable in this study,can provide reference for pharmacokinetic study and bioequivalence evaluation of cefdinir preparations.Test preparation and reference preparation are equivalent.
PBPK model;Cefdinir;Pharmacokinetics;Gastrointestinal absorption;Bioequivalence
R978.1
A
1001-0408(2016)35-4920-04
2016-01-13
2016-04-21)
(编辑:张元媛)
国家自然科学基金资助项目(No.81473287)
*硕士研究生。研究方向:药动学及药物相互作用。电话:027-85726073。E-mail:wangnx_union@163.com
#通信作者:主任药师。研究方向:药动学及药物相互作用。电话:027-85726073。E-mail:luyn_union@163.com
DOI10.6039/j.issn.1001-0408.2016.35.08
