5-苯基-2H-1,2,3-三唑-4-羧酸的合成
2016-12-30付晓滨陈云舟李欢袁华陈云峰
付晓滨,陈云舟,李欢,袁华,陈云峰
武汉工程大学化学与环境工程学院,湖北武汉430074
5-苯基-2H-1,2,3-三唑-4-羧酸的合成
付晓滨,陈云舟,李欢,袁华,陈云峰*
武汉工程大学化学与环境工程学院,湖北武汉430074
提出了两种高效的合成5-苯基-2H-1,2,3-三唑-4-羧酸的方法.首先利用三氯化铝催化的苯甲醛与硝基化合物及叠氮化钠的三组分反应高效合成含有甲基或酯基的4,5-二取代的NH-1,2,3-三唑,然后再利用两种方式得到最终目标化合物,一是利用酯基三唑水解后酸化;二是通过甲基三唑氧化,两种方法的总收率分别达到78%和61%.所得到的化合物及中间体都通过了核磁,质谱进行了表征,其波谱数据与化合物结构吻合.两种新的合成方法均具有原料易得、价格低廉、合成路线短、操作简单、总体收率高等特点,为后续该化合物的应用提供了研究基础.
三氯化铝催化;三组分反应;1,2,3-三唑;合成
1 引言
1 ,2,3 -三唑类化合物是一类重要的氮杂环化合物,三唑具有很强的生物活性,良好的生物相容性以及稳定性,在医药中间体和配位化学方面有重要的应用.1,2,3-三唑环是β-内酰胺[1]、抑菌剂[2]、止痛剂[3]等药物的良好药效基团,也是重要的抗肿瘤[4]、抗癫痫[5]等医药的重要中间体.NH-1,2,3-三唑通过N-烃基化反应,可以区域选择性的合成各种2-取代的1,2,3-三唑,因此也是极具价值的有机合成中间体[6].1,2,3-三唑环上含有连续的三个氮原子,环上电子云密度大,易于与金属离子配位,环与环之间的π-π堆积作用也可以影响到配合物的骨架结构,因此可以产生多种多样的配位模式.羧酸含有两个氧原子,配位能力强,配位方式多样[7],羧基取代的1,2,3-三唑化合物是构筑金属配合物的良好配体.
NH-1,2,3-三唑-4-羧酸也是一种重要医药中间体,可应用于溶血磷脂酸受体拮抗剂[8]、食欲肽受体拮抗剂[9]、抗炎药物[10]的合成.中山大学陈小明课题组对1,2,3-三唑-4,5-二羧酸系列的配合物做了许多研究[11-14].然而,仅含一个羧基的1,2,3-三唑目前还少见报道.2015年,本课题组报道了一种以5-苯基-2H-1,2,3-三唑-4-羧酸为配体的双核铜配合物[Cu2(H2O)2(DMF)2(L)2],并对其DNA切割活性进行了研究[15].文中涉及到5-苯基-2H-1,2,3-三唑-4-羧酸的合成,是该化合物唯一一条已见报道的合成路线.这条路线中,合成经历Sonogashira偶联、“点击”反应、酯基水解三步,收率不高.得益于本课题组对“一锅法”合成1,2,3-三唑的研究[16],这里提出两种5-苯基-2H-1,2,3-三唑-4-羧酸的合成方法.方法一是先合成带酯基的三唑,再将酯基水解后酸化;方法二是先合成带甲基的三唑,再将甲基氧化.本文提出的两种方法均具有原料易得、价格低廉、合成路线短、操作简单、总体收率较高等特点.
2 实验部分
2.1 仪器与试剂
主要仪器:Varian 600 MHz和400 MHz核磁共振仪(TMS做内标);RY-1型熔点仪(温度计未校正);FINNIGAN TRACE MS和Bruker Apex IV FTMS质谱仪.
所用试剂均为分析纯,无需重新纯化直接使用.
2.2 合成三唑方法一
第一步是一锅法合成4-苯基-5-乙酸乙酯基-2H-1,2,3-三唑(2a),第二步是2a酯基的水解与酸化,得到目标化合物3,见图1.对两步的反应条件进行了优化,结果如表1、表2所示,选出了最优条件.
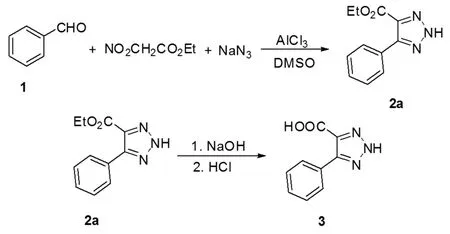
图1 5-苯基-2H-1,2,3-三唑-4-羧酸的合成方法一Fig.1Approach 1 for synthesis of 5-phenyl-2H-1,2,3-triazole-4-carboxylic acid

表1 一锅法反应条件的优化表Tab.1Optimization of reaction conditions for one-pot synthesis

表2 4-苯基-5-乙酸乙酯基-2H-1,2,3-三唑水解条件的优化表Tab.2Optimization of hydrolysis of ethyl 5-phen-2H-1,2,3-triazole-4-carboxylate
2.2.1 化合物2a的合成首先,对合成2a的条件进行了优化,确定了最优反应条件为以AlCl3为催化剂,DMSO为溶剂,温度为70℃.如表1所示,对下列苯甲醛与硝基乙酸乙酯及NaN3的反应,(NaN3在酸性以及金属存在下容易发生爆炸,使用时注意安全.)采用非质子型溶剂DMSO,控制反应温度和溶液浓度相同,在不同的Lewis催化剂FeCl3、ZnBr2、AlCl3,AlCl3催化效果最佳.确定催化剂为AlCl3,以20℃为间隔调整反应温度,确定70℃为最佳反应温度.考虑溶剂对反应效果的影响和溶剂处理的难易程度,控制其它变量,尝试了DMF和CH3CN作为反应溶剂,但是收率并不理想.另外,还增加了对反应液浓度的控制,发现当反应液的浓度(以硝基乙酸乙酯的浓度为标准)控制在0.2 mmol/mL的时候,最利于反应收率的提高.降低浓度,反应收率改变不大,却增加了溶剂的用量;升高浓度,由于副反应增多,中间产物硝基烯烃容易聚合,反应收率下降.
具体实例:向100 mL圆底烧瓶中加入苯甲醛500 mg(4.72 mmol),加入20 mL DMSO稀释苯甲醛溶液,再向其中滴加硝基乙酸乙酯753 mg(5.67 mmol);称取NaN3460 mg(7.08 mmol),倒入烧瓶中;搅拌上述混合液体,加入94 mg(0.7 mmol)黄色AlCl3粉末.上述混合溶液置于80℃油浴锅中,恒温回流反应,用TLC监测反应进程.反应结束后,将混合溶液倒入水中,用乙酸乙酯/水萃取3次;将有机层合并,用饱和食盐水洗涤一次;乙酸乙酯层用无水硫酸钠干燥,抽滤后,减压下脱去乙酸乙酯;再用(洗脱剂)乙酸乙酯∶石油醚(1∶3)进行柱分离,得到产物淡黄色固体2a(871 mg,收率85%).m.p.:90℃~92℃.1HNMR(400MHz,CDCl3) δ:7.87-7.77(m,2H),7.49-7.38(m,3H),4.38(q,J=7.2 Hz,2H),1.31(t,J=7.2 Hz,3H);13C NMR(100 MHz,CDCl3)δ:161.2,129.7,129.3,128.3,127.7,61.7,14.1.MS(EI):m/z217.[17]
2.2.2 化合物2a的水解与酸化表2是4-苯基-5-乙酸乙酯基-2H-1,2,3-三唑水解条件优化.首先,控制温度80℃,回流时间5 h,分别采用10 mL、20 mL、30 mL 0.5 mol/L的NaOH溶液,发现NaOH用量不足将会降低水解收率,而较多的NaOH对收率提高影响不大.其次,10℃为一个梯度,尝试降低水解反应的温度,发现60℃时,水解反应收率也可以达到92%,继续降低温度则会导致水解不完全.随后,对反应时间进行了优化,缩短反应时间收率降低,延长反应时间,收率增加不显著.最终确定的最佳水解条件为20 mL 0.5 mol/L NaOH,在60℃条件下回流5 h.
具体实例:取871 mg化合物2a,加入20 mL 0.5 mol/L NaOH,加热到60℃,TLC监测反应进程.反应结束后,再用1 mol/L HCl将溶液的pH值调节到2,再用乙酸乙酯/水体系萃取3次,有机层合并,并用无水硫酸钠干燥,抽滤后,减压下脱去乙酸乙酯,得到最终产物淡黄色固体3(699 mg,收率92%).m.p.:200℃~202℃.1H NMR(600 MHz,d6-DMSO)δ:7.83(d,J=6.6 Hz,2H),7.52-7.30(m,3H);13C NMR(150 MHz,d6-DMSO)δ:162.1,131.4,129.2,129.0,128.2.MS(EI):m/z189.
2.3 合成三唑方法二
第一步是采用一锅法合成5-甲基-4-苯基-2H-1,2,3-三唑(2b),第二步是通过对2b的甲基进行氧化得到目标产物3,见图2、表3.
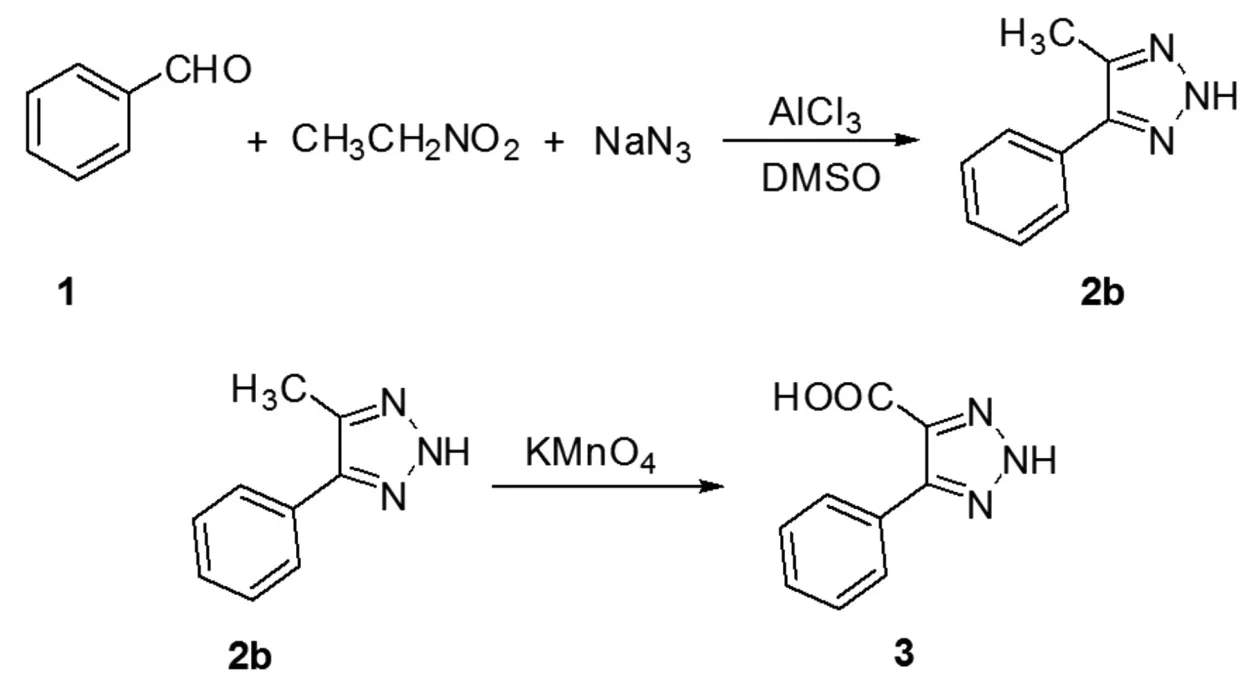
图2 5-苯基-2H-1,2,3-三唑-4-羧酸的合成方法二Fig.2Approach 2 for synthesis of 5-phenyl-2H-1,2,3-triazole-4-carboxylic acid

表34 -甲基-5-苯基-NH-1,2,3-三唑氧化反应条件的优化表Tab.3Optimization of oxidation reaction of 4-methyl-5-phenyl-NH-1,2,3-triazole
2.3.1 化合物2b的合成参照化合物2a的合成最佳条件.向100 mL圆底烧瓶中加入苯甲醛500 mg(4.72 mmol),用20 mL DMSO稀释苯甲醛溶液,再向其中滴加硝基乙烷426 mg(5.67 mmol);称取NaN3460 mg(7.08 mmol),倒入烧瓶中;加94 mg(0.7 mmol)黄色AlCl3粉末.上述混合溶液置于80℃油浴锅中,恒温回流反应,反应进程主要采用TLC监测.反应结束后,将混合溶液倒入水中,用乙酸乙酯/水萃取3次;将有机层合并,用饱和食盐水洗涤1次;乙酸乙酯层用无水硫酸钠干燥,抽滤后,减压下脱去乙酸乙酯;再用(洗脱剂)乙酸乙酯∶石油醚(1∶5)进行柱分离,得到产物淡黄色固体2b(721 mg,收率96%).m.p:165℃~166℃.1H NMR(600MHz,DMSO-d6):δ:7.73-7.28(m,5H),2.42(s,3H);13C NMR(150 MHz,DMSO-d6)δ:132.2,129.2,127.1,11.9.HRMSCalcd(ESI)m/zfor C9H10N3:[M+H]+160.0869,found:160.0874.[18]
2.3.2 化合物2b的氧化得到化合物2b后,以高锰酸钾为氧化剂,优化了高锰酸钾的用量及反应温度,最佳反应条件为:高锰酸钾用量为3 mol/L,氧化温度80℃,反应时间5 h.控制温度为80℃,反应时间为4 h,改变氧化剂的用量,发现采用3 mol/L KMnO4最合适,KMnO4用量不够反应收率不理想,继续增加氧化剂用量,对反应效果无显著提升.控制反应温度和时间,10℃为一个梯度,试验温度对反应的影响,发现温度偏高和偏低都会导致收率下降,尤其是温度较低时,甲基无法完全氧化成羧基.继续对氧化反应的时间进行优化,发现4 h已经是最适宜的反应时间,反应时间不够将会使氧化不够完全,导致收率偏低.
具体实例:取化合物2b(721 mg,4.53 mmol)全部倒入瓶中,再称取2.15 g(13.59 mmol)KMnO4放到100 mL圆底烧瓶中混合,加入40 mL蒸馏水;上述混合溶液放到75℃恒温油浴锅中,通过TLC进行监测.反应结束后,抽滤,除去反应瓶中的固体,并且用水洗涤固体3次.洗涤和抽滤的液体合并后,用1 mol/L HCl调节溶液的pH值到2,减压下脱去大部分水,加入适量的甲醇,加热溶解后重结晶,得到最终产物3(548 mg,收率64%).
3 结果与讨论
已报导的化合物5-苯基-2H-1,2,3-三唑-4-羧酸的合成方法仅有一例[12].其合成分为三步:(1)Sonogashira偶联;(2)“点击”反应;(3)酯基水解. 3步总收率52%,见图3.
该方法的弊端主要在于:(1)路线包含3步,总体收率偏低;(2)Sonogashira偶联反应的原料为碘苯,该物质价格昂贵,且不稳定;偶联反应需要用到钯催化剂,价格昂贵,且反应条件需要无氧,操作上也比较麻烦.

图3 5-苯基-2H-1,2,3-三唑-4-羧酸的合成Fig.3Synthesis of 5-phenyl-2H-1,2,3-triazole-4-carboxylic acid
本文提出的两种合成路线突出的优点是:原料易得、价格低廉、合成路线短、操作简单、总体收率较高.具体分析如下:
对于方法一,第一步是以苯甲醛为原料,通过“一锅法”合成1,2,3-三唑[13].这一步简化了文献中的第一步和第二步反应,有效的避开了繁琐的操作以及昂贵、不稳定的试剂和催化剂,且反应的收率非常可观,可以达到85%.该路线的第二步,存在的问题也是原子经济性不高;但与方法二相比,优点是转化率高,收率高,第二步酯基水解的反应,收率可以达到92%.两步收率为78%.
对于方法二,由于“一锅法”为合成1,2,3-三唑提供了非常简便、快捷、高效的方式,我们可以先合成4-苯基-5-甲基-2H-1,2,3-三唑,再将三唑上的甲基用KMnO4氧化为羧基,从而合成目标产物.方法二的整体收率为61%.该方法的主要优势在于,所用原料苯甲醛、硝基乙烷均为价格低廉的化学试剂;缺点是,相对于方法二,反应需要的时间较长,甲基的氧化不容易完全.
4 结语
本报道提供了两种5-苯基-2H-1,2,3-三唑-4-羧酸的合成方法,并且分析了不同方法的优缺点.利用苯甲醛与硝基化合物及叠氮化钠的三组分反应高效合成4,5-二取代的NH-1,2,3-三唑,再通过酯基的水解与酸化,或者甲基的氧化,可以高效合成5-苯基-2H-1,2,3-三唑-4-羧酸,保留后续该化合物及其衍生物的合成和性质研究有重要参考价值.
[1]KUZIN A P,NUKAGA M,NUKAGA Y,et al. InhibitionoftheSHV-1β-lactamasebysulfones:crystallographicobservationoftworeaction intermediates with tazobactam[J].Biochemistry,2001,40(6):1861-1866.
[2]KARANAM M,DEV S,CHOUDHURY A R.New polymorphs of fluconazole:results from cocrystallization experiments[J].Crystal growth&design,2012,12(1):240-252.
[3]BRATSOS I,URANKAR D,ZANGRANDO E,et al.1-(2-Picolyl)-substituted1,2,3-triazoleasnovel chelatingligandforthepreparationofruthenium complexes with potential anticancer activity[J].Dalton trans,2011,40(19):5188-5199.
[4]FISHER S Z,AGGARWAL M,KOVALEVSKY A Y,et al.Neutron diffraction of acetazolamide-bound human carbonic anhydrase II reveals atomic details of drug binding[J].Journal of the american chemistry society,2012,134(36):14726-14729.
[5]GALL M,KAMDAR B V,COLLINS R J.Pharmacology of some metabolites of triazolam,alprazolam,and diazepam prepared by a simple,one-step oxidation of benzodiazepines[J].Journal of medicinal chemistry,1978,21(12):1290-1294.
[6]张文生,匡春香,杨青.NH-1,2,3-三唑合成进展[J].有机化学.2011,31(1):54-62.
ZHANG W S,KUANG C X,YANG Q.Progress for the synthesis of NH-1,2,3-triazole[J].Chinese journal of organic chemistry,2011,31(1):54-62.
[7]李艳秋.新型稀土二元羧酸配合物及稀土苯甲酸衍生物配合物[D].北京:首都师范大学,2008.
[8]BUCKMANBO.Lysophosphatidicacidreceptor antagonists:WO 2013025733A1[P].2013-02-21.
[9]ZHANG Z.Hydrazonopyrazole deravtives and their use as therapeutics:WO 03045379[P].2003-06-05.
[10]BENJAMIN P.New triazole compounds useful in the treatmenr of inflammation:WO 2008135767A1[P]. 2008-11-13.
[11]ZHANGWX,XUEW,LINJB,et al.3D geometricallyfrustratedmagnetsassembledby transitionmetalionand1,2,3-triazole-4,5-dicarboxylateastriangularnodes[J].Crystal engineering communications,2008,10(12):1770-1776.
[12]ZHANG W X,XUE W,CHEN X M.Flexiblemixedspin Kagomé coordination polymers with reversible magnetism triggered by dehydration and rehydration[J].Inorganic chemistry,2011,50(1):309-316.
[13]YUAN G,SHAO K Z,DU D Y,et al.Syntheses,structures,and photoluminescence of d10coordination architectures:from 1D to 3D complexes based on mixed ligands[J].Solid state sciences,2011,13(5):1083-1091.
[14]SHI W,CHEN X Y,XU N,et al.Synthesis,crystal structures,and magnetic properties of 2D manganese(II)and 1D gadolinium(III)coordination polymers with 1H-1,2,3-triazole-4,5-dicarboxylic acid[J]. Europe journal of inorganic chemistry,2006(23):4931-4937.
[15]LIU W Q,ZHOU S L,FAN M Z,et al.Synthesis and crystal structure of a dinuclear Cu(II)complex based on a carboxyl-substituted 1H-1,2,3-triazole and its DNAcleavageactivity[J].Chinesejournalof structure chemistry,2015,34(6):917-924.
[16]HU Q,LIU Y,DENG X,et al.Aluminium(III)chloride-catalyzedthree-componentcondensationof aromatic aldehydes,nitroalkanes and sodium azide for thesynthesisof4-aryl-NH-1,2,3-triazoles[J]. Advanced synthesis&catalysis,2016,358(10):1689-1693.
[17]CHASIH,GUOR,YINW,etal.One-pot,three-componentreactionusingmodifiedJulia reagents:a facile synthesis of 4,5-disubstituted 1,2,3-(NH)-triazoles in a wet organic solvent[J].ACS combinatorial science,2015,17(3):147-151
[18]QUAN X,REN Z,WANG Y.p-Toluenesulfonic acid mediated 1,3-dipolar cycloaddition of nitroolefins with NaN3for synthesis of 4-aryl-NH-1,2,3-triazoles[J]. Organic letters,2014,16(21):5728-5731.
本文编辑:张瑞
Synthesis of 5-phenyl-2H-1,2,3-triazole-4-carboxylic acid
FU Xiaobin,CHEN Yunzhou,LI Huan,YUAN Hua,CHEN Yunfeng
School of Chemistry and Environmental Engineering,Wuhan Institute of Technology,Wuhan 430074,China
Two high efficient methods of synthesis of 5-phenyl-2H-1,2,3-triazole-4-carboxylic acid were developed.Firstly,4,5-substitued-NH-1,2,3-triazole bearing methyl or ester groups were synthesized by using AlCl3-catalyzed three-component reaction of benzaldehyde,nitroalkane and sodium azide,then the target products were obtained via two methods:one is the hydrolysis and acidification of ester-substituted 1,2,3-triazole,the other is the oxidation of methyl-substituted 1,2,3-triazole,and the overall yields by these two methods were up to 78%and 61%respectively.The structures of the target compound and relative intermediates were confirmed by proton nuclear magnetic resonance spectroscopy,carbon-13 nuclear magnetic resonance spectroscopy and mass spectrometry.It is demonstrates that the methods have the characteristics of easy available raw materials,low cost,short synthetic route,simple operation and high overall yield,which provides the research basis for the application of this compound.
AlCl3-catalyzed;three-component reaction;1,2,3-triazole;synthesis
TQ252.6
A
10.3969/j.issn.1674-2869.2016.06.003
1674-2869(2016)06-0527-05
2016-10-10
武汉科技大学煤转化与新型炭材料湖北省重点实验室基金(WKDM201502);武汉工程大学研究生创新基金(CX2015160)
付晓滨,助理工程师.E-mail:jojopyne@163.com
*通讯作者:陈云峰,博士,教授.E-mail:chyfch@hotmail.com
