MISPE-HPLC检测水产品5种丁香酚麻醉剂的残留量
2016-12-29何洪健
何洪健
黄 和1,2,3
高 平4
刘 亚1,2,3
蒋志红1,2,3JIANG Zhi-hong1,2,3
曹湛慧1,2,3
(1. 广东省水产品加工与安全重点实验室,广东 湛江 524088;2. 广东普通高等学校水产品深加工重点实验室,广东湛江 524088;3. 广东海洋大学食品科技学院,广东 湛江 524088;4. 湛江市食品药品检验所,广东 湛江 524022)
MISPE-HPLC检测水产品5种丁香酚麻醉剂的残留量
何洪健1,2,3
黄 和1,2,3
高 平4
刘 亚1,2,3
蒋志红1,2,3JIANGZhi-hong1,2,3
曹湛慧1,2,3
(1. 广东省水产品加工与安全重点实验室,广东 湛江 524088;2. 广东普通高等学校水产品深加工重点实验室,广东湛江 524088;3. 广东海洋大学食品科技学院,广东 湛江 524088;4. 湛江市食品药品检验所,广东 湛江 524022)
釆用分子印迹固相萃取结合高效液相色谱法,建立检测水产品中5种丁香酚类麻醉剂残留的方法。分别使用丁香酚、甲基丙烯酸、乙二醇二甲基丙烯酸酯作为模板分子、功能单体和交联剂,采用本体聚合法制备合成分子印迹聚合物并制成萃取柱。通过响应面分析法得出模板分子、功能单体、交联剂的最佳比例1∶4.55∶23.21;样品溶剂选择甲醇能使5种丁香酚类化合物回收率达到90%;洗脱剂选择9∶1的甲醇∶乙酸溶液能够使5种丁香酚类化合物回收率达90%以上;该方法的线性范围均为 0.05~10 μg/mL,相关系数为 0.999 83~0.999 97,具有较好的线性关系;5种丁香酚类麻醉剂的定量限为0.05 mg/kg。该方法对样品的回收率为82.04%~99.29%,RSD为0.06%~3.96%。所建立的方法具有简单便于操作、准确快速、操作可行性高、有机溶剂耗量少等特点,可用于水产品中5种丁香酚类麻醉剂残留的测定。
分子印迹聚合物;分子印迹固相萃取;高效液相色谱法;水产品;丁香酚类麻醉剂
分子印迹技术(molecular imprinting technique,MIT)是指为获得在空间结构和结合位点上与某一目标化合物(模板分子或印迹分子)完全匹配的高分子聚合物的制备技术,由于具有特异识别性、构预性和广泛适用性三大优势,被广泛运用于医药、食品科学和环境等领域[1]。分子印迹聚合物(Molecularly imprinte polyers,MIPs)是人工合成的,对特定分子具有特异选择性的聚合物,MIPs的制备和应用是分子印迹技术的核心。分子印迹固相萃取(molecular imprinted soild phase extraction,MISPE)与传统的固相萃取相比,利用MIPs的特性选择性,将其用于高效液相(high performance liquid chromatography,HPLC)色谱固定相具有广泛的应用前景,既可拆分手性物质,也可分离常规固定相难以分离的相似物;既可应用于定性定量分析,也可用于制备性分离[2]。
丁香酚类化合物作为鲜活水产品的麻醉剂的安全性一直受质疑。美国食品药品监督管理局(FDA)也禁止丁香酚化合物作为活鲜食用水产品的麻醉剂和镇定剂;有些国家如澳大利亚、新西兰、智利等也严格规定了其休药期和残留量。而中国至今并没有相关部门正式审批丁香酚类渔用麻醉剂应用于水产品中的规定。因此,亟需建立水产品中多种丁香酚类化合物的高效、准确的样品前处理方法和仪器定量、验证方法。
现阶段,中国检测丁香酚类化合物的方法有高效液相色谱法[3-5]、液质联用[6-8]、气相色谱法[9]、气质联用[10]、荧光光谱法[11]、毛细管电泳高频电导法[12]、紫外分光光度法[13]等,前处理主要有微波辅助萃取、分散固相萃取[14]等,这些方法的不足之处主要对含有多种丁香酚类化合物的基质样品和复杂的基质样品检测还有一定的困难。本试验拟釆用分子印迹固相萃取技术和高效液相色谱法,建立一种检测水产品中5种丁香酚类麻醉剂残留的方法,对MIPs的制备以及其固相萃取的重要因素(模板分子、功能单体、交联剂、萃取洗脱剂、淋洗剂等)进行合理系统的优化,旨在为水产品中丁香酚类麻醉剂的检测提供试验依据。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂
南美白对虾、石斑鱼、鳜鱼:湛江市霞山区昌大昌超市;
丁香酚标准品(CAS号:97-53-0)、异丁香酚标准品(CAS号:97-54-1)、甲基丁香酚标准品(CAS号:93-15-2)、甲基异丁香酚标准品(CAS号:93-16-3)、乙酰基异丁香酚标准品(CAS号:93-29-8):纯度在98%以上,成都艾科达化学试剂有限公司;
偶氮二异丁腈、乙二醇二甲基丙烯酸酯、α-甲基丙烯酸:分析纯,成都艾科达化学试剂有限公司;
甲醇:色谱纯,天津市四友精细化学品有限公司;
乙腈:色谱纯,德国 Merch 公司。
1.1.2 主要仪器设备
高效液相色谱仪:1260型,美国Agilent 公司;
组织匀浆机:HR2860型,珠海飞利浦有限公司;
漩涡混合仪:XW-80A型,海门市其林贝尔仪器制造有限公司;
超声波清洗器:HS-10260D型,北京天地首和科技发展有限公司;
紫外分光光度计:TU1901型,北京普析通用仪器有限责任公司;
电子分析天平:XS-205型,美国Mettler-Toledo公司;
超纯水机:arium-611VF型,德国Sartorius公司;
氮吹仪:N-EVAP111型,美国Organomation公司;
数显恒温水浴锅:HH-6型,常州国华仪器有限公司;
高速离心机:3-18K型,德国Sigma公司。
1.2 试验方法
1.2.1 分子印迹固相萃取柱的制备 分子印迹萃取柱制备流程是将模板分子(丁香酚)、功能单体(甲基丙烯酸)、交联剂(乙二醇二甲基丙烯酸酯)按照一定的比例混合,振荡摇匀后静置30 min,然后加入1 mmol的引发剂偶氮二异丁腈(EGDMA),超声处理(800 W,5 min),氮吹15 min后密封,恒温振荡水浴55 ℃,加热24 h取出聚合物,研磨过筛,收集40~80 μm的颗粒。以甲醇—乙酸(体积比9∶1)混合溶剂为洗脱剂,使用索氏洗脱法洗脱40 h,改用甲醇洗脱8 h将乙酸洗掉,至洗脱液在分光光度计检测不到丁香酚。非分子印迹聚合物与分子印迹聚合物制备流程相同,但未加模板分子。称量200 mg聚合物置于装有0.45 μm的筛板的2 mL 注射器中,压实后再次压入筛板。以乙腈和高纯水作为淋洗剂淋洗萃取柱,真空抽干后备用。分别用3 mL高纯水和乙腈加压淋洗,再依次用2 mL甲醇—乙酸(体积比9∶1) 混合溶剂和高纯水淋洗,真空泵抽干约2 min,备用。
1.2.2 样品前处理 称取5.00 g样品,置于50 mL离心管中,加入15 mL提取溶剂甲醇溶液,涡旋振荡混匀1 min,然后用超声波清洗器超声提取5 min,功率为800 W,放入高速离心机中9 500 r/min离心5 min,吸取上层清液,剩下的残渣再用15 mL甲醇溶液按照上述步骤混匀、超声、离心、吸取上清液,合并两次上清液;以3 mL甲醇—乙酸(体积比9∶1)混合溶剂和超纯水作为淋洗剂淋洗活化萃取柱,取30 mL待测样品溶液以7~9 mL/min的流速过萃取柱,此后,加入3 mL的高纯水进行淋洗并且用真空泵抽干,使用2 mL甲醇—乙酸(体积比9∶1)混合溶剂以0.5 mL/min的流速对萃取柱进行洗脱,收集洗脱液在40 ℃氮气保护下吹干,加入1.0 mL流动相甲醇溶液溶解。再将上述溶液过0.22 μm的有机滤膜到进样小瓶,供高效液相色谱分析。
1.2.3 响应面试验 在温度为55 ℃,引发剂为1 mmol EGDMA,模板洗脱使用甲醇—乙酸(体积比9∶1)混合液进行索氏洗脱法洗脱,以回收率作为因变量,考察功能单体、模板分子以及交联剂比例对丁香酚分子印迹聚合物效果的影响并找到最佳方案。
1.2.4 静态吸附试验 称取200 mg的MIPS分别置于装有25 mL质量浓度在 0~30 mg/mL范围丁香酚溶液的50 mL离心管中,涡旋振荡5 min,以4 500 r/min的转速离心10 min,然后移入150 mL烧杯,置于摇床在25 ℃、61 r/min条件下振摇24 h,使用分光光度计检测吸附后的浓度,并按式(1)计算吸附量,考察其吸附效果。
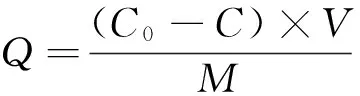
(1)
式中:
Q——聚合物在吸附平衡时的吸附量,mg/g;
C0——吸附溶液吸附前质量浓度,mg/mL;
C——吸附溶液吸附后质量浓度,mg/mL;
V——吸附溶液的体积,mL;
M——分子印迹聚合物的质量,g。
1.2.5 分子印迹固相萃取柱的优化 通过单因素试验,考察不同样品溶剂、洗脱剂、淋洗剂对分子印迹固相萃取的效果。
(1) 样品溶剂对分子印迹固相萃取效果的影响:样品加标浓度为5 mg/kg,采用甲醇、乙酸乙酯、乙腈等作为提取溶剂,按照1.2.2 的流程进行样品处理,高效液相检测并计算其回收率。
(2) 淋洗剂对分子印迹固相萃取效果的影响:样品加标浓度为5 mg/kg,采用乙腈、甲醇、水作为淋洗液,按照1.2.2 的流程进行样品处理,高效液相检测并计算其回收率。
(3) 洗脱液对分子印迹固相萃取效果的影响:乙酸、甲醇、乙腈、氯仿以及甲醇—乙酸(体积比9∶1)混合液作为洗脱液,加标浓度为5 mg/kg,按照1.2.2 的流程进行样品处理,高效液相检测并计算其回收率。
1.2.6 高效液相色谱条件 色谱柱:C18柱(250 mm×4.6 mm,5 μm);柱温:35 ℃;流动相体积比:甲醇∶乙腈∶水=24∶24∶52;流速:1.0 mL/min;检测器:紫外检测器;检测波长:280 nm;进样量:20 μL。
2 结果与分析
2.1 分子印迹聚合物的制备
2.1.1 分子模板、功能单体、交联剂比例的选择 试验中分子印迹聚合物采用本体聚合法作为制备的方法,在温度为55 ℃、引发剂为1 mmol EGDMA的条件下,参照文献[15]引入了多元变量分析技术考察各参数的最优值,通过曲线响应面法考察模板、单体以及交联剂比例对分子印迹聚合物回收效果的影响。根据单因素优化结果进行试验因素水平的选取,使用Design-Expert软件进行多响应值的最优条件选择,试验设计及结果见表1~3 。
以丁香酚的回收率作为响应值,根据表3的结果分析,拟合回归方程,剔除不显著项得到各因子对回收率影响的回归方程:
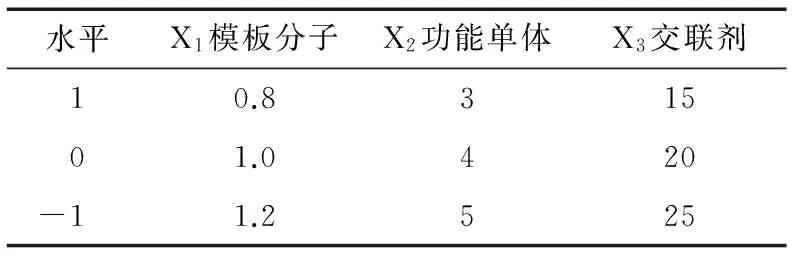
表1 响应面分析因素与水平Table 1 The response surface analysis of factors and levels mmol
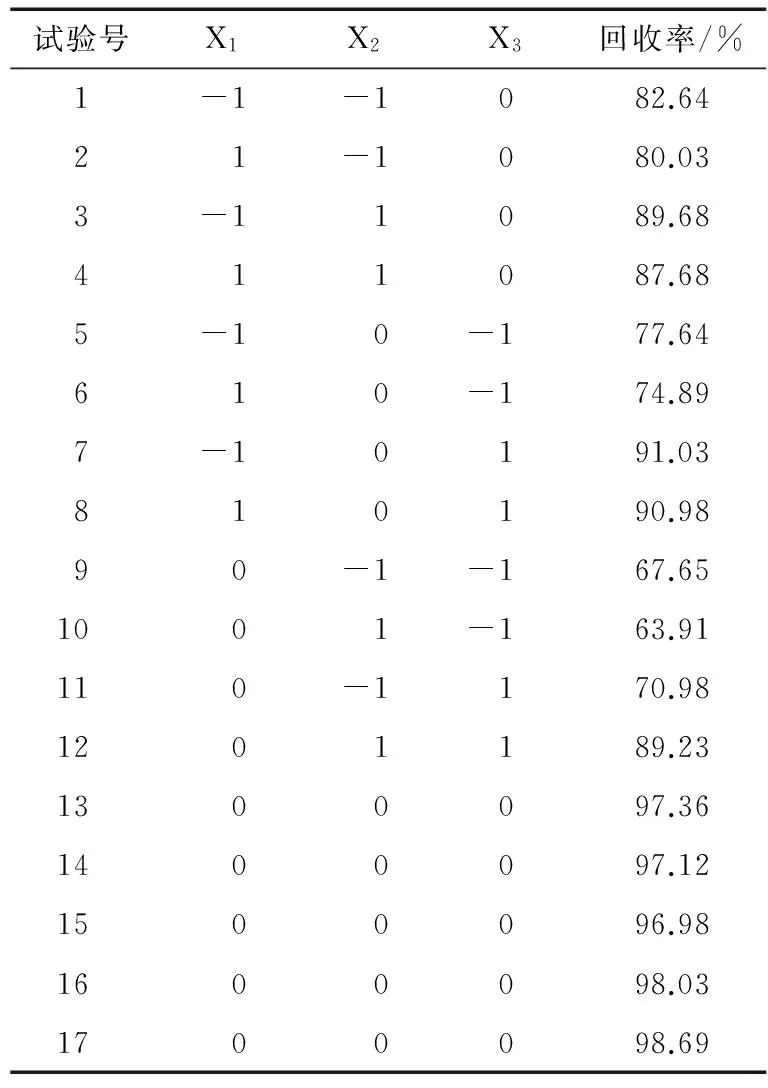
表2 响应面分析方案及试验结果Table 2 The response surface analysis and the experimental results
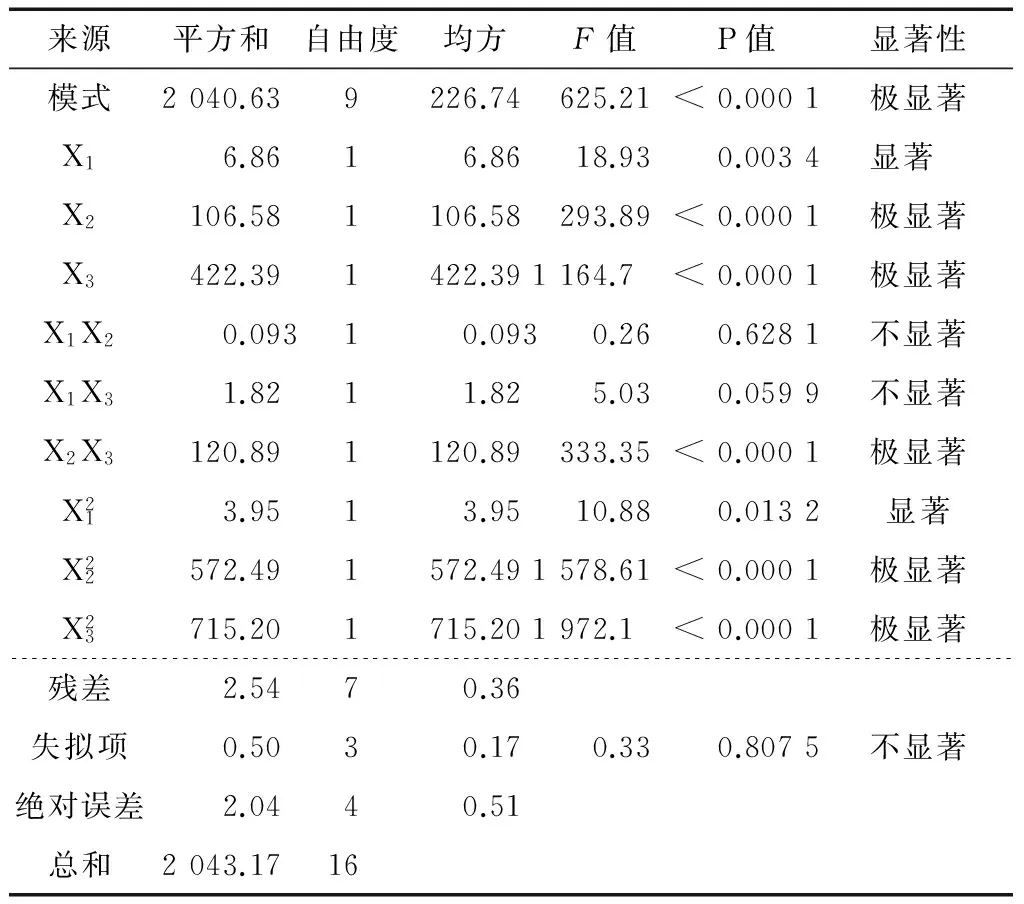
表3 回归结果分析†Table 3 Regression result analysis
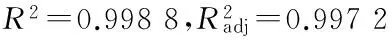
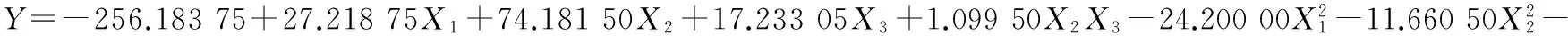
(2)

使用Design-Expert软件进行多响应值的最优条件选择,得到最佳聚合反应条件为:模板分子0.92 mmol,功能单体4.23 mmol,交联剂21.59 mmol,即三者最佳比例为1∶4.55∶23.21。
根据模板分子∶功能单体∶交联剂=1∶4.55∶23.21,1 mmol模板分子、4.55 mmol功能单体、23.21 mmol交联剂、1 mmol引发剂、15 mL二氯甲烷制备丁香酚分子印迹聚合物,用浓度为10 mg/kg的浓度丁香酚标准溶液进行过柱萃取,结果表明,丁香酚标准液回收率为98.28%,RSD为1.29%,说明该方案可行。

图1 模板分子、功能单体以及交联剂比例的结果示意图
Figure 1 Template molecule and functional monomer and crosslinking agent proportion of results
2.1.2 分子印迹聚合物吸附效果的考察 借鉴袁列江等[16]对分子印迹聚合物吸附性能的考察方式,通过静态吸附试验考察分子印迹聚合物以及非分子印迹聚合物对丁香酚溶剂的吸附效果,试验分别取浓度范围为0~35 mg/mL的丁香酚溶液,考察相同浓度下两种聚合物的不同吸附效果。由图2可知,分子印迹聚合物的吸附量随着丁香酚溶液浓度的增加而快速增大;而非分子印迹聚合物的吸附量增加平缓。在相同的丁香酚溶液浓度下,分子印迹聚合物的吸附量均大于非分子印迹聚合物的吸附量,这是因为在分子印迹聚合物中含有能够与模板分子在特殊形状、一定尺寸以及功能基团相匹配的三维空穴,使得MIPs对于模板分子丁香酚具有特异性结合的作用,因此MIPs吸附能力得到较大提高。另一方面则是非分子印迹聚合物是由功能单体的随机聚合,对丁香酚类物质没有特异性吸附的能力,所以吸附能力相对较差。
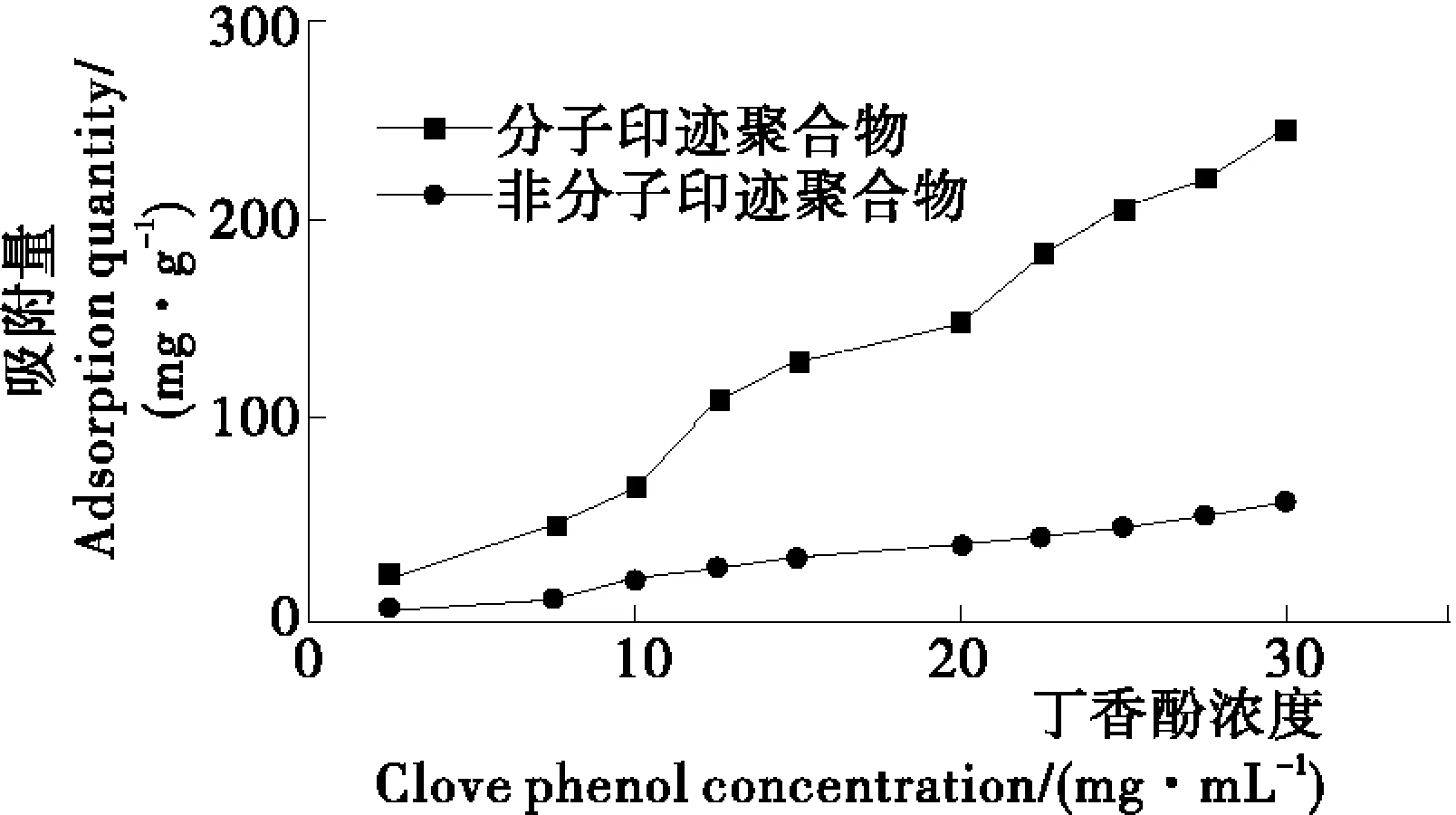
图2 两种分子聚合物中的吸附效果图
Figure 2 The adsorption effect of two kinds of molecular weight polymer
2.2 分子印迹固相萃取的优化
2.2.1 样品溶剂对吸附效果的影响 借鉴肖琴等[17]对分子印迹固相萃取优化的方法,考察溶剂对回收率的影响。丁香酚、异丁香酚、甲基丁香酚、甲基异丁香酚、乙酰基异丁香酚都是溶于极性的有机溶剂,根据文献[18~19],通常采用甲醇、乙酸乙酯、乙腈等作为提取溶剂。本试验中考察甲醇、乙酸乙酯、乙腈作为样品溶剂,结果(图3)表明选择乙酸乙酯作为样品溶剂分子印迹萃取柱的吸附率最低,乙腈作为样品溶剂吸附率在67.10%~88.13%,而甲醇标准溶液进行分子印迹固相萃取,对于5种溶质的吸附率在87.03%~95.46%,可能是以丁香酚为模板分子,各种样品溶剂中的丁香酚吸附率都相对较高。
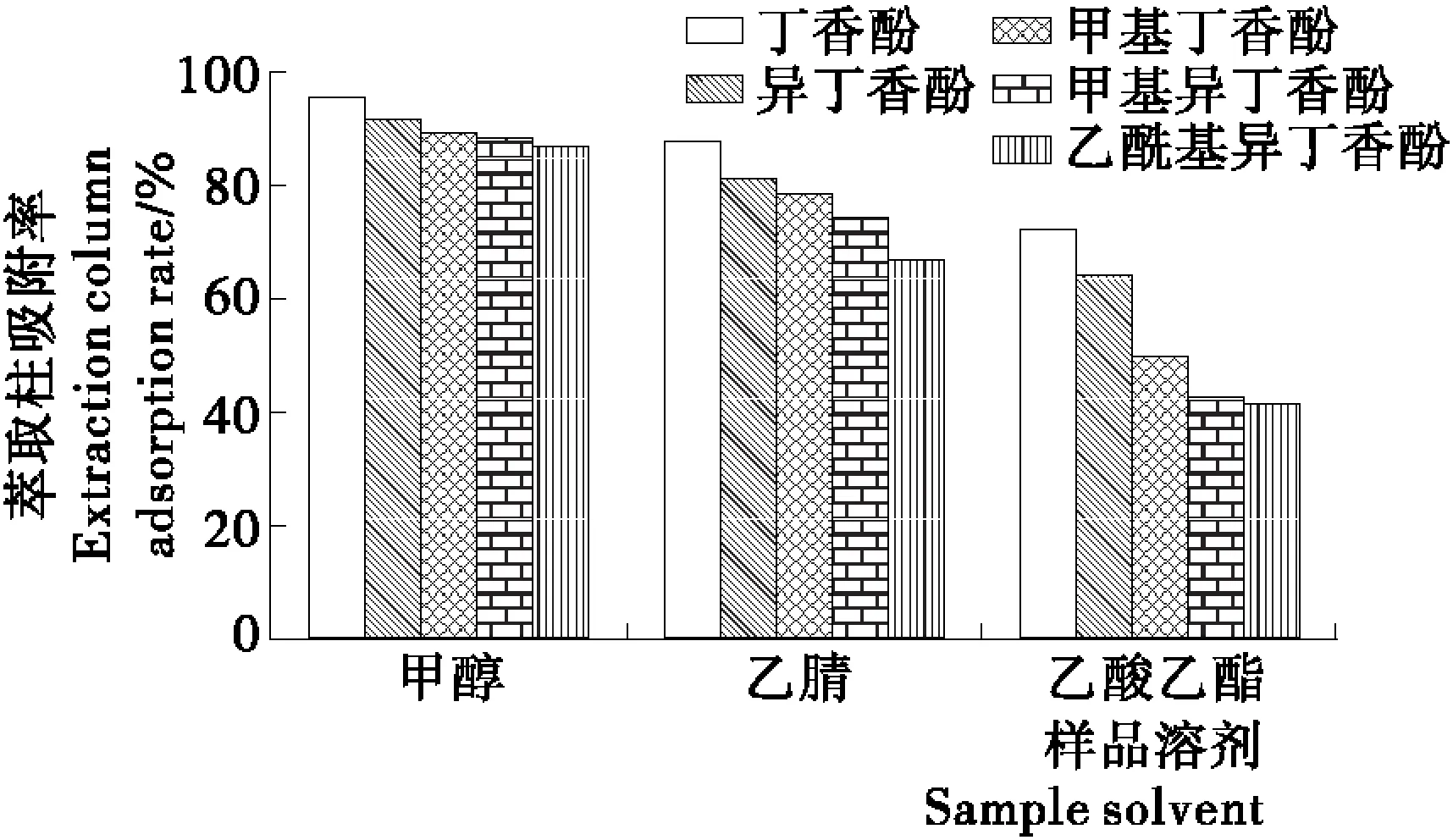
图3 不同样品溶剂的吸附效果Figure 3 The effect of different sample solvents
2.2.2 淋洗液的选择 本试验考察乙腈、甲醇、水作为淋洗液,试验中,提取加标浓度为5 mg/kg的石斑鱼样品,过柱,考察不同的淋洗液对回收率的影响。结果(表4)表明,乙腈、水作为淋洗液时回收率都能达到90%以上,而甲醇作为淋洗液回收率最低,根据进一步考察在淋洗过程中使用水和乙腈分别进行淋洗,可以在保证回收率的同时,最大限度去除杂质,达到提纯净化的目的。
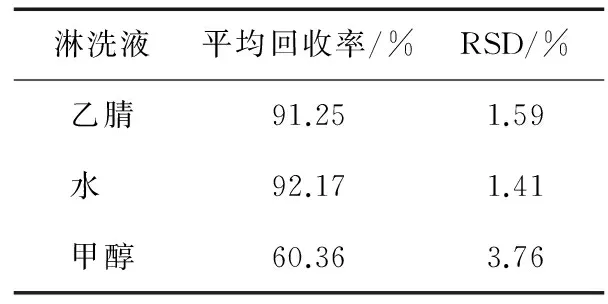
表4 不同淋洗液的淋洗效果Table 4 The effect of different leacheates (n=3)
2.2.3 洗脱剂种类的影响 洗脱溶剂能有效地从柱上解吸附,这对目标化合物的纯化和富集具有很大的作用,因此,选择适合的洗脱剂对萃取柱的萃取效果十分重要。本试验主要考察乙酸、甲醇、乙腈、氯仿以及甲醇—乙酸(体积比9∶1)混合液作为洗脱剂对萃取柱洗脱效果的影响。由图4可知,甲醇和甲醇—乙酸混合液的效果都比较好,但是甲醇—乙酸混合液效果相对较好,丁香酚、异丁香酚、甲基丁香酚、甲基异丁香酚、乙酰基异丁香酚的总量分别占分子迹固相萃取柱吸附总量的102.32%,98.46%,90.37%,91.97%,90.67%。出现这种情况主要是因为底物的键合作用较弱,使用甲醇做为洗脱剂能一定程度上防止聚合物在洗脱剂中溶胀、缩小,并且乙酸使得其酸度适度增加,这样能使其与分析物的结合能力增强,因此,选用甲醇—乙酸(体积比9∶1)混合液作为洗脱剂。
2.3 分子印迹固相萃取柱对样品净化和检测效果
分别使用C18柱、MISPE柱对石斑鱼加标浓度为1.20 mg/kg样品溶液进行萃取,过滤膜进样。由图5、6可知,经C18柱萃取后石斑鱼样品色谱图杂峰较多,富集净化作用不明显,而经MISPE柱萃取的石斑鱼样品色谱图杂峰较少,峰形较好,基线噪音较少。由表5可知,C18柱与MISPE柱的萃取平均回收率分别为91.82%、54.47%,MISPE柱回收率相当于C18柱的两倍左右。出现这种现状的主要原因是MISPE柱对丁香酚类化合物具有特异性结合的功能,对其净化和富集效果明显,能有效地用于水产品中的丁香酚类麻醉剂的测定。

图4 不同洗脱剂的洗脱效果Figure 4 The effect of different eluents表5 不同萃取柱的净化效果Table 5 Extraction effects of different extraction column

萃取柱种类平均回收率/%RSD/%杂质干扰峰形基线噪音C1854.471.43~3.62较多较好较小MIPPE柱91.820.45~2.31较少较好较小

1. 丁香酚 2. 异丁香酚 3. 甲基丁香酚 4. 甲基异丁香酚 5. 乙酰基异丁香酚
图5 石斑鱼C18柱的液相色谱图
Figure 5 Grouper C18column liquid chromatogram(1.20 mg/kg)

1. 丁香酚 2. 异丁香酚 3. 甲基丁香酚 4. 甲基异丁香酚 5. 乙酰基异丁香酚
图6 石斑鱼过分子印迹固相萃取柱液相色谱图
Figure 6 Grouper molecularly imprinted solid-phase extraction column liquid chromatogram
2.4 线性关系与检测限
用有机流动相甲醇分别配制成不同浓度(0.05,0.10,0.20,0.50,1.00,5.00,10.00 μg/mL)的混合标准溶液,然后按照设定的仪器条件进行HPLC分析,以浓度(x,μg/mL)为横坐标,以峰面积(y)为纵坐标作图,分别绘制5种丁香酚类麻醉剂的标准曲线,结果(表6)显示:5种丁香酚类麻醉剂在0.05~10.00 μg/mL浓度范围内,线性关系良好,相关系数均高于0.999 0。按照标准样品的上机条件,取10 g阴性石斑鱼样品,进行提取、富集、洗脱、浓缩、定容,当进样量为20 μL时,以10倍信噪比(S/N)计算定量限,该方法对5种丁香酚类麻醉剂的定量限(LOQ)均为0.05 mg/kg。标样浓度为10.0 mg/L的液相色谱图见图7。
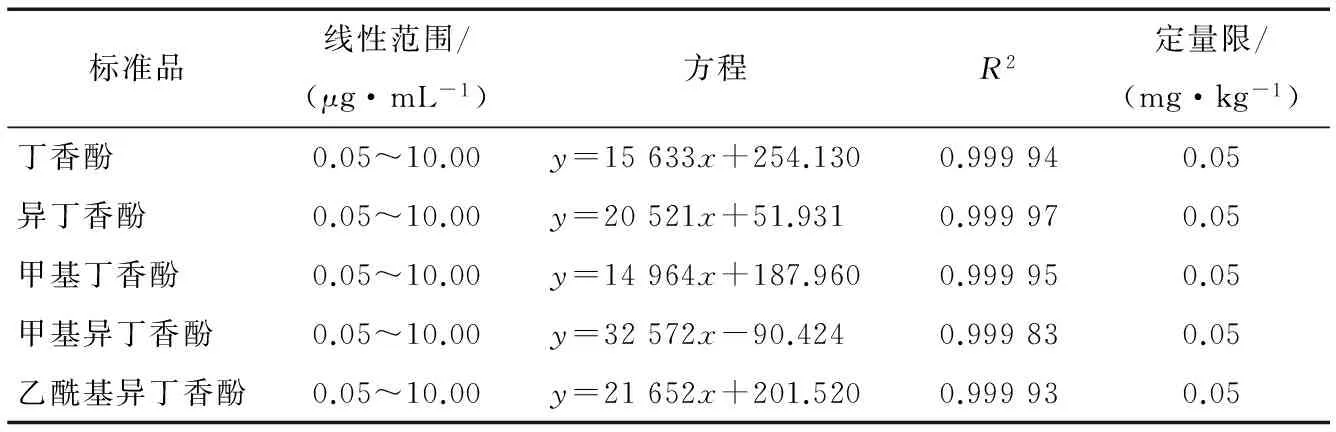
表6 5种丁香酚类麻醉剂标准品线性方程、R2、定量限Table 6 Five clove pheno drug standard linear equation and R2, the detection limit
2.5 方法的重现加标回收率和精密度
向空白石斑鱼、桂花鱼、南美白对虾肉样品中添加0.20,0.50,1.00 mg/kg 3个浓度梯度的混合标准溶液,对其进行加标回收率试验测定,得出的结果见表7。由表7可知,5种丁香酚类麻醉剂平均加标回收率为82.04%~99.29%,其相对标准偏差值为0.06%~3.96%,说明本方法的重现性好,同时符合药物残留的检测要求。
3 结论
本试验旨在以分子印迹固相萃取作为样品前处理技术,与高效液相色谱法相结合,建立一种可用于水产品中丁香酚类麻醉剂残留检测的新方法。系统研究了影响分子印迹固相萃取的因素如模板分子、功能单体、交联剂的比例、样品溶剂、洗脱剂等,并对方法的分析性能进行了综合评价。在优化条件下,丁香酚、异丁香酚、甲基丁香酚、甲基异丁香酚、乙酰基异丁香酚5种分析物均有较高的灵敏度,样品的加标回收率为 82.04%~99.29%,RSD在0.06%~3.96%。综上所述,该方法简单快速,有机溶剂用量少,可应用于水产品中丁香酚类麻醉剂残留的测定,但是分子印迹固相萃取的聚合物制备方法以及萃取的模式还需要作更多的尝试和更深入的研究。

1. 丁香酚 2. 异丁香酚 3. 甲基丁香酚 4. 甲基异丁香酚 5. 乙酰基异丁香酚
图7 280 nm波长下的5种丁香酚吸收图谱
Figure 7 280 nm wavelength of five kinds of eugenol absorption spectrum (10.0 μg/mL)

表7 5种丁香酚类化合物加标回收率和相对标准偏差Table 7 The standard addition recoveries and relative standard deviation of five kinds of clove phenol compounds (n=5)
[1] 王颖, 李楠. 分子印迹技术及其应用[J]. 化工进展, 2010, 29(12): 2 315-2 323.
[2] 卢春阳, 何海成, 何锡文, 等. 除草剂青芳定分子印迹聚合物的合成及结合性能研究[J]. 化学学报, 2004, 62(8): 799-803.
[3] 卢智玲, 刘华栋. 反相高效液相色谱法测定克痢痧胶囊中丁香酚的含量[J]. 中国卫生检验杂志, 2011, 21(9): 2 190-2 191.
[4] 余小平. RP-HPLC 法测定中药丁香中丁香酚的含[J]. 中华中医药学刊, 2009, 27(4): 880-881.
[5] 陈焕, 黄和, 高平, 等. 高效液相色谱法测定水产品中丁香酚类麻醉剂的残留量[J]. 食品与发酵工业, 2014, 40(12): 156-160.
[6] 张忠盼. 液-液萃取-液相色谱-串联质谱法测定“地沟油”中辣椒碱类化合物及丁香酚[J]. 色谱, 2012, 30(11): 1 108-1 112.
[7] 芦智远, 刘辰乾, 冯歆轶, 等. 超高效液相色谱-串联质谱法鱼肉丁香酚的残留量测定[J]. 科学养鱼, 2016, 47(7): 76-77.
[8] 赵秀丽, 陈大为, 任飞,等. LC/MSn鉴定丁香酚布洛芬酯在大鼠尿中的主要代谢产物[J]. 中国药学杂志, 2005, 40(21): 66-69.
[9] 王哲民, 魏学兵. 气相色谱法测定藏药静心胶囊中丁香酚的含量[J]. 中成药, 2006, 28(10): 1 536-1 538.
[10] 周相娟, 江改青, 李伟. 气相色谱-质谱法测定香水中的甲基丁香酚[J]. 分析试验室, 2013, 32(5): 104-106.
[11] 赵文武, 刘翠格, 魏永巨. 丁香酚的荧光光谱及其在中药丁香质量评价中的应用[C]// 第十五届全国分子光谱学术报告会论文集. [出版地不详]:中国光学学会, 中国化学学会, 2008: 327-328.
[12] 林伟丰, 吴小林, 陈缵光, 等. 毛细管电泳高频电导法测定几种中药中的丁香酚[J]. 高等学校化学学报, 2006, 27(11): 2 070-2 072.
[13] 张智敏, 朱倩倩. 紫外分光光度法测定丁香油和风油精中丁香酚的含量[J]. 安徽工业大学学报: 自然科学版, 2006, 23(3): 272-274.
[14] 陈焕, 黄和, 高平, 等. 分散固相萃取-气相色谱法同时测定水产品中六种丁香酚类麻醉剂的残留量[J]. 食品工业科技, 2015, 36(8): 89-92.
[15] KOOHPAEI A R, SHAHTAHERI S J, GANJALIC M R, et al. Application of multivariate analysis to the screening of molecularly imprinted polymers(MIPs) for ametryn[J]. Talanta, 2008, 75: 978-986.
[16] 袁列江, 王会娟, 李忠海, 等. 双酚A磁性分子印迹聚合物的制备及性能研究[J]. 食品与机械, 2015, 31(2): 93-97.
[17] 肖琴, 林一, 郑琳, 等. 分子印迹固相萃取—高效液相色谱法检测鱼肉中的孔雀石绿和结晶紫[J].粮油食品科技, 2011, 19(6): 31-34.
[18] 李永红, 王瑞, 孙志浩. RP-HPLC 方法同时测定生物转化液中的香草醛和异丁香酚含量[J]. 河南工业大学学报: 自然科学版, 2007, 28(5): 50-52.
[19] 李得恩. 保健食品中丁香酚的毛细管气相色谱测定法[J]. 环境与健康杂志, 2008, 25(7): 574.
Determination of five kinds of clove phenol anesthetic in aquatic product by HPLC with molecularity imprinted solid phase extraction
HE Hong-jian1,2,3
HUANGHe1,2,3
GAOPing4
LIUYa1,2,3
CAOZhan-hui1,2,3
(1.GuangdongProvincialKeyLaboratoryofAquaticProductProcessingandSafety,Zhanjiang,Guangdong524088,China; 2.KeyLaboratoryofAdvancedProcessingofAquaticProductsofGuangdongHigherEducationInstitution,Zhanjiang,Guangdong524088,China; 3.CollegeofFoodScienceandTechnology,GuangdongOceanUniversity,Zhanjiang,Guangdong524088,China;4.ZhanjiangInstituteofSupervision&TestonFood&Medicines,Zhanjiang,Guangdong524022,China)
A new method of molecularly imprinted solid phase extraction (MISPE) combined with high performance liquid chromatography (HPLC) was developed for detecting five kinds of clove phenol anesthetic in aquatic product. Using eugenol as template molecule, methacrylic acid as functional monomer, ethylene glycol dimethacrylate as cross linking agent, molecularly imprinted polymers were synthesized by bulk polymerization and made into extraction column. Through the response surface analysis template molecule and functional monomer, crosslinking agent the best proportion was 1∶4. 55∶23. 21, and the recovery rate of these compounds reached 90% by using methanol as solvent and acetic acid solution as eluent. The results showed that linear response data were obtained in the concentration range of 0.05~10 μg/mL for the five kinds of clove phenol anesthetic with correlation coefficients from 0.999 83 to 0.999 97, and the limits of quantitative were 0.05 mg/kg. The method of sample recovery rate was 82.04%~99.29% with the relative standard deviations ranging from 0.06%~3.96%. The new established method was found simple and fast,accurate and feasible, and this could be successfully applied for the determination of five kinds of clove phenol anesthetic in aquatic product and their residue with low solvent consumption.
Molecularly imprinted polymer; molecularly imprinted solid phase extraction (MISPE) ; high performance liquid chromatography (HPLC); aquatic product; clove phenol anesthetic
广东省科技计划项目(编号:2014A020208121)
何洪健,男,广东海洋大学在读硕士研究生。
黄和(1962—),男,广东海洋大学教授。 E-mail:zjhahe@163.com
2016—06—02
10.13652/j.issn.1003-5788.2016.11.009
