分子间相互作用研究的APF-D密度泛函SAM色散校正方法改进
2016-12-29王一波
何 禹 王一波,*
(1贵州省高性能计算化学重点实验室,贵阳550025;2贵州大学网络与信息中心,贵阳550025)
分子间相互作用研究的APF-D密度泛函SAM色散校正方法改进
何 禹1,2王一波1,2,*
(1贵州省高性能计算化学重点实验室,贵阳550025;2贵州大学网络与信息中心,贵阳550025)
APF密度泛函色散校正(APF-D)是由B3PW91和PBE0杂化的APF泛函,加上球形原子模型(SAM)色散校正构成的一种有别于Grimme经验色散校正的密度泛函理论色散校正(DFT-D)方法,计算稀有气体及其与小分子复合物结合能和势能面准确性很高,但对常见氢键、C―H…π和π…π等复合物结合能计算结果明显偏大,在一般性分子间相互作用问题研究中一直未被认可和采纳。我们发现APF-D结合能计算结果偏大的原因是APF泛函与SAM色散校正重复计入了一定量的长程色散能;通过引入不依赖于体系的SAM色散能阻力因子ζ,简单而有效地解决了APF-D色散能过度补偿问题,提出了APF-D改进方案APF-D*;通过S66和L7标准测试集的对照计算表明,APF-D*结合能准确性远高于原始APF-D,达到和超过目前常用的B3LYPD3和ωB97X-D方法水平,并具有较好的计算效率,期待在大体系分子间相互作用研究中得到广泛应用。
DFT-D;APF泛函;SAM色散校正
Key Words:DFT-D;APF functional;SAM dispersion correction
1 引言
快速、精确计算分子间相互作用能,准确预测其复合物几何结构是对量子化学计算方法的重要挑战1。目前常用的近似密度泛函方法由于缺乏对长程色散作用的有效描述,用于分子间相互作用问题的研究显得没有单分子体系计算那样出色2-6。Grimme研究组7长期致力于发展近似密度泛函方法的经验色散校正模型,近年来提出的DFTD3取得重要进展,特别是传统B3LYP泛函结合D3色散校正的B3LYP-D3在分子间相互作用领域得到广泛认可。此外Head-Gordon研究组发展的长程分离杂化泛函ωB97X结合色散校正的ωB97X-D方法8,早于B3LYP-D3被较多研究者所采纳9。
2012年,著名量子化学程序Gaussian的主要开发者Frisch、Petersson等提出了Austin-Petersson-Frisch泛函,简称APF,它由41.1%B3PW91和58.9%PBE0泛函混合而成,加上球形原子模型(SAM)经验色散校正构成APF-D方法,也称APFPFD(Petersson-Frisch-Dispersion)10,并完成了一、二阶解析能量梯度程序化,发布在Gaussian 09 Release D.01/E.01的标准方法中11。APF-D的作者用此方法计算了稀有气体双原子、稀有原子与小分子复合物的结合能和几何结构,获得了与CCSD (T)/aug-cc-pVTZ计算可比拟的精确结果10。在计算效率上,由于构成APF的B3PW91和PBE0均属于Hybrid泛函,计算量与Hartree-Fock SCF在同一量级,对双电子积分和网格精度也不苛求,计算速度远高于ωB97X-D和M05/M06等Minnesota系列泛函。令人遗憾的是,APF-D发表至今已近五年时间,一直未能进入研究分子间相互作用密度泛函方法的主流行列。
DFT-D方法计算结果的准确性除了DFT泛函和色散校正模型自身的品质外,很大程度取决于两者对色散作用描述的互补性或契合程度12;同时,在发展DFT-D方法时优化相关组合参数所依赖的训练集也很重要13。我们用Hobza的分子间相互作用标准测试集S6614,以APF-D/aug-cc-pVTZ逐一计算各体系的结合能,对照CCSD(T)/CBS“金标准”时发现,对于氢键、C―H…π,π…π和C―H…C―H等常见类型的体系,APF-D方法结合能计算结果平均偏大12%左右(见Supporting Information(SI)中Table S3),这点也曾被原作者在论文展望部分提及10,但迄今尚未见进一步的研究工作发表。不难发现,这种误差起源于确定APF-D组合参数所使用的训练集,偏重选择结合强度很弱的稀有气体van der Waals作用,无论何种DFT泛函自身都难以很好地描述这类弱作用,构造DFT-D时,只好依赖经验色散来补偿,对于氢键、C―H…π和π…π相互作用,这种尺度确定的经验色散补偿就显得过度了。
为了修正这种过度的补偿,我们使用S66标准测试集,通过大量优化、统计分析,提出了APFD色散校正方法的改进方案APF-D*,对S66和L7测试集15系统地进行了测试、分析,同时还研究了APF-D*方法的基函数效应,发现Pople的6-311++ G(2d,p)作为与APF-D*配合使用的基函数是相对准确、高效的选择。在计算效率方面,以L7测试集的大体系为例,在提出DFT-D方法原始的训练集基函数下,APF-D*方法具有较高的计算效率。
2 方法与计算
2.1APF-D色散能分析
苯-奈复合物有S、T和Pd三种构型(图1),分别代表了π…π、C―H…π和两者混合类型的相互作用,准确计算它们的结合能并保持正确的大小顺序,对于各类计算方法都具有挑战性。最近我们组完成了该体系的CCSD(T)/aug-cc-pVQZ结合能计算,评估了常用于分子间相互作用研究的多种密度泛函方法16,本文以此体系为例来探讨和分析APF-D方法色散能组成所存在的问题。
APF-D计算获得的色散能ΔEAPF-Ddisp由APF泛函提供的ΔEAPFdisp和借助SAM球原子模型获得的经验色散ΔESAMdisp两部分构成:

ΔEAPFdisp主要包括了短程、中程色散作用,同时也有少量长程色散,ΔESAMdisp则以长程色散为主。
在超分子近似下,色散能与相关能虽是不同概念,但色散能从数值上与结合能中的电子相关能贡献项大致相等,基于Morokuma能量分解方案发展起来的计算方法已广泛接受和应用了这一近似17,即:


图1 苯-奈复合物S、T和Pd三种构型Fig.1 Sandwich,T-shaped,and parallel-displaced configurations of benzene-naphthalene complex
ΔEAPFcorr可表达为APF结合能ΔEAPF与同名基函数下获得的Hartree-Fock结合能ΔEHF之差:

由此可得:
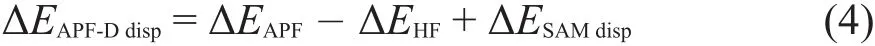
本文计算的三种构型苯-奈复合物ΔEAPF-Ddisp与高精度CCSD(T)/aug-cc-pVQZ相关能ΔECCSD(T)corr列于表1,不难发现,ΔEAPF-Ddisp比CCSD(T)结果大10%-12%,APF泛函计算出的ΔEAPFdisp与源于SAM经验色散的ΔESAMdisp之和重复计入了一定量的长程色散作用,使得总色散能被夸大。
有两种办法可修正ΔEAPF-Ddisp色散能被过度补偿,一种是保持SAM经验色散能ΔESAMdisp不变,优化泛函,减少APF泛函中的长程色散作用部分,另一种则是保持APF泛函不变,减少SAM经验色散的ΔESAMdisp数值。由于组成APF泛函的B3PW91在中、长程距离表现出一致的排斥,而在相同范围PBE0表现出一致的吸引10,41.1%B3PW91和58.9%PBE0的组合能有效去除混合泛函容易产生的负面影响,APF泛函是可信赖的,本文保持它不变,着力修正SAM色散项。我们使用经测试集优化、统计分析找到的阻尼因子ζ乘上ΔESAMdisp,扣除长程色散的重复部分,修正总体色散能得到改进的APF-D*方案。即:
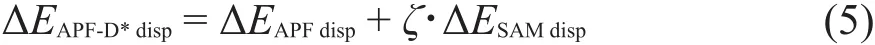
2.2APF-D*方法阻尼因子ζ的确定
S22测试集是Hobza研究组提出,早期被广泛使用的分子间相互作用的测试集18-20,但是其体系较少,只有22对复合物,代表性不够强。我们选择了包括66对复合物,涵盖了典型的化学、生物和材料分子间相互作用的S66测试集作为样本来确定阻尼因子ζ。S66测试集对静电和色散作用的考量较为均衡,其中氢键组选择了23对典型的不同强度和类型的以静电作用为主的氢键复合物,色散组选择了23对涵盖π…π、C―H…π和C―H…C―H等以色散作用为主的复合物,混合组选择了20对静电与色散并存的复合物。其中各复合物的几何结构数据是用校正BSSE的MP2/cc-pVTZ方法优化得到,结合能数据采用计算化学“金标准”——CCSD(T)/CBS标准方法计算结果14。
APF-D方法在S66测试集下表现出SAM模型对长程色散作用的过度补偿,改进APF-D方法的关键是消除SAM模型与APF泛函重复计入的长程色散项。从S66测试集的统计结果(图3与SI附件Table S1)可以看出,对测试集中的每种复合物,APF-D方法都表现出色散作用的夸大,导致总结合能一致性的系统偏大,而且夸大的幅度也大致相近,这使得改进APF-D的办法变得简单易行,没有必要去寻找依赖体系的阻尼函数,对每一体系采用不同尺度的校正,而是SAM色散项乘上一个经过优化的统一的阻尼因子常数即可,这点对APF-D改进方案的准确度至关重要。
对于各种方法与CCSD(T)/CBS计算结果差别的统计分析,使用了平均偏差(MD)、平均绝对偏差(MAD)与均方根偏差(RMSD)统计指标21(定义见SI附件)。其中RMSD最重要,反映了数据整体质量好坏和偏差的离散程度,MD与MAD反映了系统的误差,通常使用RMSD与MAD综合评价,MD偏差涉及偏差正负抵消,仅作参考。
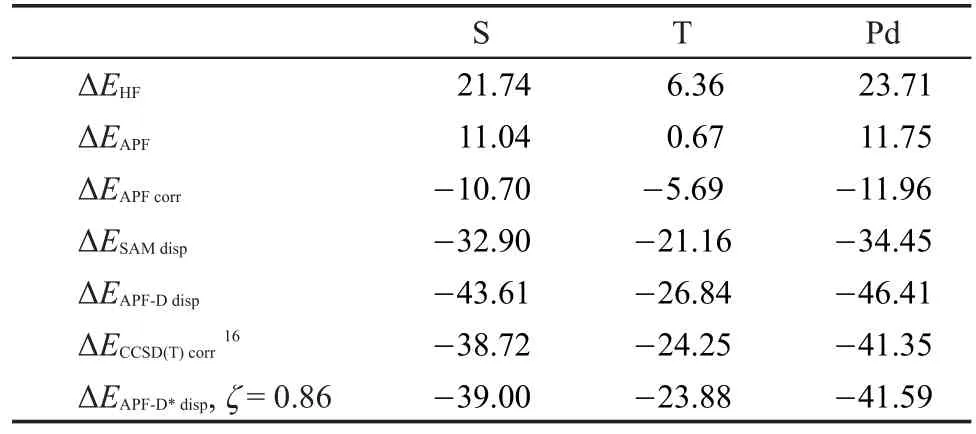
表1 使用aug-cc-pVQZ基组计算S、T和Pd三种构型苯-奈复合物结合能(kJ·mol-1)分解Table 1 Bind energy(kJ·mol-1)decompositions for different configurations of benzene-naphthalene complex using aug-cc-pVQZ basis set
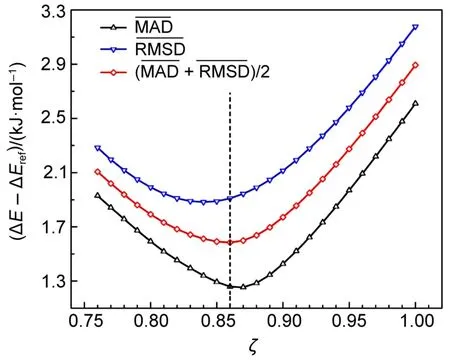
图2 阻尼因子(ζ)与APF-D*方法结合能偏差的相关性Fig.2 Correlation of resistance factor(ζ)and the bind energies ofAPF-D*method
具体确定阻尼因子ζ的步骤为:首先采用APFD方法,分别选择6-311G(2d,p)、6-311++G(2d,p)、6-311++G(2d,2p)、cc-pVDZ、cc-pVTZ、aug-ccpVDZ、aug-cc-pVTZ、def2-TZVP、def2-TZVPP和def2-QZVP等10种基函数,对S66测试集的复合物进行660次计算,将结合能分解为APF泛函结合能与SAM色散能。然后设定ζ在0.75至1之间,以0.01为步长递增取值,用ζ校正SAM色散能,针对每种基函数计算出APF-D*与CCSD(T)参考标准值的MAD与RMSD偏差值,再计算出10种基函数的MAD与RMSD的平均值与,最后以指标与的算术平均值极小来确定最优ζ值(详见SI附件Table S2)。
2.3 计算过程
为了分析APF-D*对APF-D方法结合能计算的改进效果,我们在众多的密度泛函中选择了B3LYP-D3和ωB97X-D方法,这两种方法是目前公认的研究分子间相互作用的优秀方法,同时基函数分别采用B3LYP-D3与ωB97X-D方法原作者推荐的def2-QZVP和6311++G(3df,3pd)基组。为研究APF-D*的基组效应、筛选与之适应的高效率基函数,我们选择了Gaussian程序中标准的6-311++ G(2d,p)和Duning22-24相关一致基组aug-cc-pVXZ (X=D,T)以及Weigend和Ahlrichs25的def2-TZVPP和def2-QZVP基组,结合能计算全部采用了均衡校正法(CP)校正了BSSE26。本文所有计算使用Gaussian 09 Release D.01程序10,在贵州省高性能计算化学重点实验室和贵州大学云计算平台的计算机集群上完成。
3 结果及讨论
3.1 APF-D*精度改善效果及其与原APF-D比较
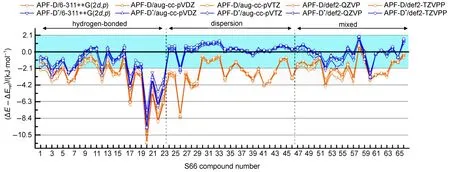
图3 多种基函数配合APF-D*与APF-D方法基于S66测试集的计算精度比较Fig.3 Comparison of calculation precision ofAPF-D*andAPF-D for different basis sets based on S66 data set
分别用APF-D和改进后的APF-D*方法结合6-311++G(2d,p)、aug-ccpVDZ、aug-cc-pVTZ、def2-TZVPP与def2-QZVP基组计算了S66测试集各复合物的结合能,以CCSD(T)/CBS为标准计算出每个点的绝对误差值,并统计出MD、MAD与RMSD值,结果见SI附件(Table S3)与表2与图3。
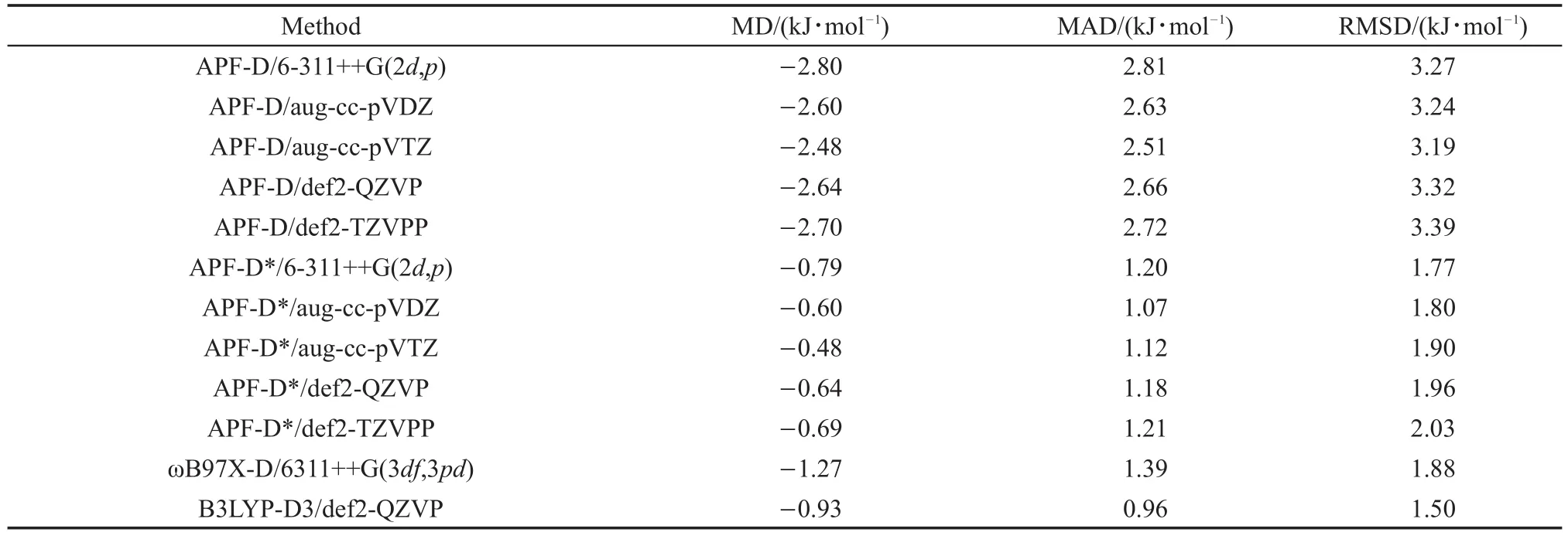
表2 基于S66测试集的用APF-D,APF-D*,ωB97X-D和B3LYP-D3方法对使用CCSD(T)/CBS基准所得的误差分析Table 2 Calculated errors ofAPF-D,APF-D*,ωB97X-D and B3LYP-D3 methods with respect to the benchmark CCSD(T)/CBS calculations on the S66 data set
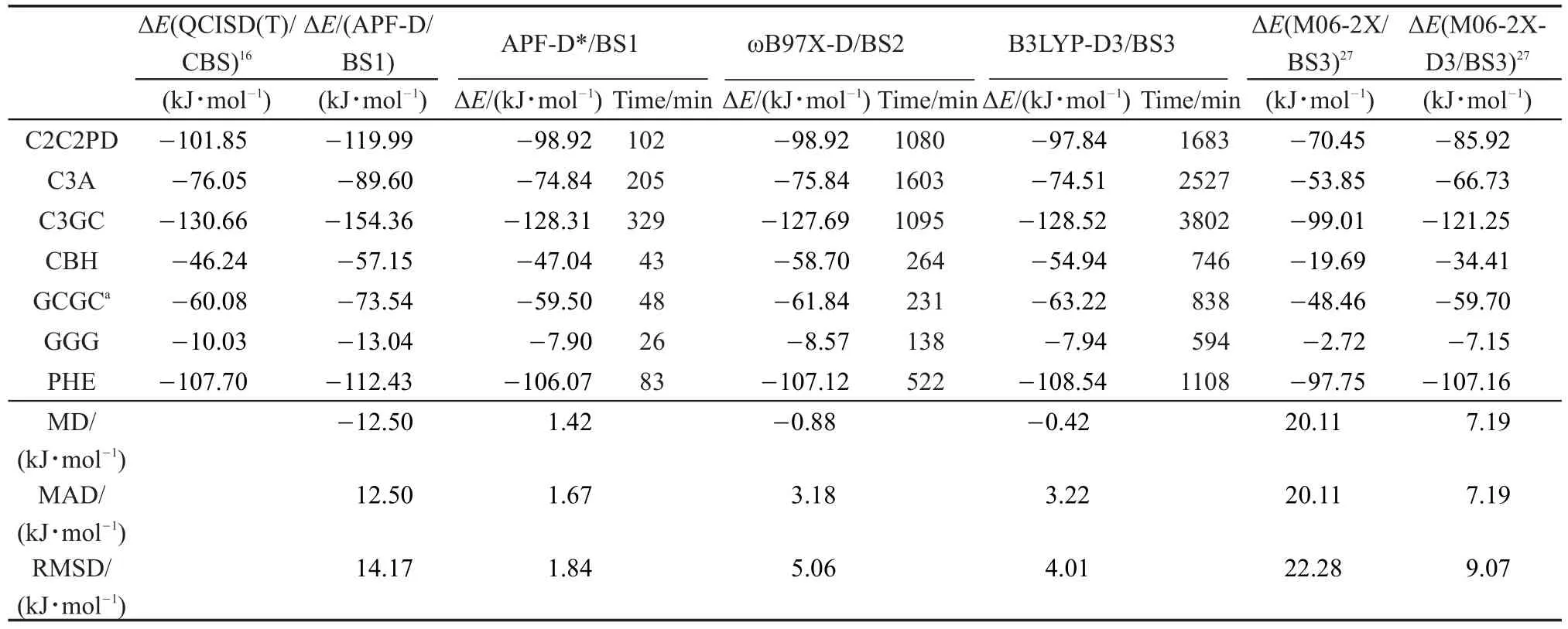
表3 基于L7测试集使用QCISD(T)/CBS基准计算所得的误差分析Table 3 Calculated errors of the studied methods with respect to the benchmark QCISD(T)/CBS calculations on the L7 data set
分析这些结果可以看到,与CCSD(T)/CBS结果比较,AFP-D方法在不同基组下结合能计算结果均不理想,结合能数值整体偏大,平均达到3 kJ· mol-1左右的误差。约70%的S66复合物结合能误差超过2.1 kJ·mol-1,其中,氢键组23个复合物只有6个误差小于2.1 kJ·mol-1,其中17、20、22和23号复合物最为明显,误差超过6.3 kJ·mol-1,最大的20号复合物误差达到-11.3 kJ·mol-1;色散组23个复合物只有6个误差小于2.1 kJ·mol-1,其余多在-4.2 kJ·mol−1左右,26号复合物误差达到-8.2 kJ·mol-1;混合组20个复合物只有8个误差小于2.1 kJ·mol-1,其余都夸大约4.2 kJ·mol-1。
改进后的APF-D*方法结合能计算误差显著降低,与CCSD(T)/CBS结果相比较,平均误差降低到1.3 kJ·mol-1以下,S66复合物误差超过2.1 kJ· mol-1的比例降低到10%左右。以色散组效果最为明显,23个复合物中22个误差小于2.1 kJ·mol-1,且很靠近基准线,特别是26号复合物由原来的-8.2 kJ·mol-1下降到-2.2 kJ·mol-1;混合组20个测试复合物有18个误差小于2.1 kJ·mol-1,剩余的2个误差也较接近2.1 kJ·mol-1;氢键组23个复合物有15个小于误差2.1 kJ·mol-1,APF-D夸大色散能最为明显的17、20、22和23号复合物均得到约3.3 kJ·mol-1的修正。说明APF-D*方法较好地修正了APF泛函与SAM色散模型重复计入长程色散能的问题。
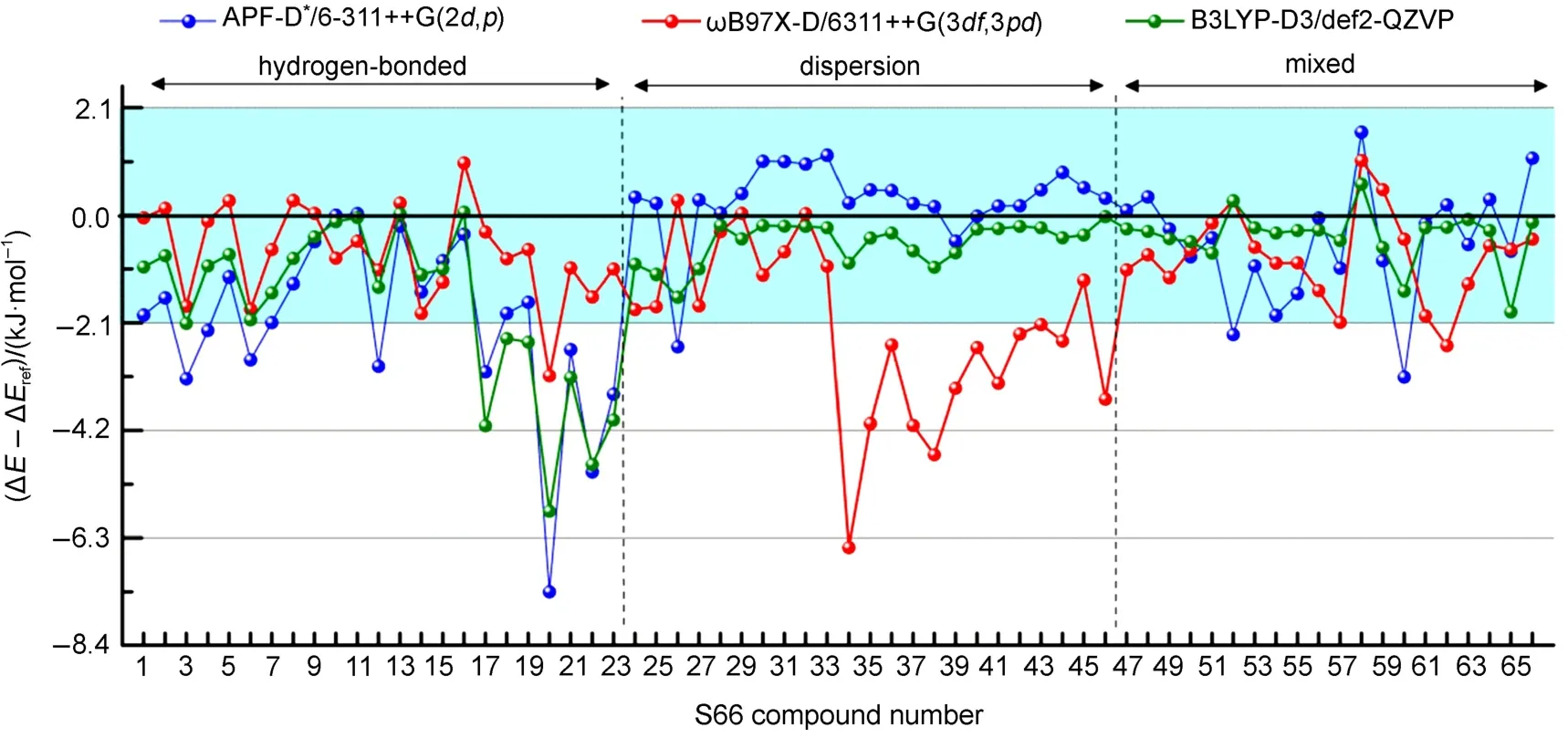
图4APF-D*与B3LYP-D3和ωB97X-D方法基于S66测试集的计算精度比较Fig.4 Comparison of calculated precision ofAPF-D*and B3LYP-D3 with ωB97X-D based on S66 data set
分析上述数据,我们还发现APF-D*结合能计算结果对基函数的依赖并不大,当然也不可无止境地使用过小基组,6-311++G(2d,p)基的MAD与RMSD值均能与更大的aug-cc-pVTZ或def2-QZVP基组相比拟,APF-D*配合6-311++G(2d,p)基是最有效的组合。
3.2APF-D*与B3LYP-D3和ωB97X-D的比较
APF-D*比APF-D有了明显的改进,但是与目前公认的分子间相互作用计算方法B3LYP-D3和ωB97X-D相比较,计算精度和计算效率如何,是它有无存在价值的重要考验,我们以S66和L7测试集进行了系统的比较。
测试计算中B3LYP-D3使用它的训练集基函数def2-QZVP,ωB97X-D选择它的训练集基函数6-311++G(3df,3pd),使得这两种方法具有最优表现,APF-D*选择6-311++G(2d,p)基函数,所有计算均校正了BSSE,以CCSD(T)/CBS为标准计算出每个点的绝对误差值。
3.2.1 S66测试集
S66测试集的MD、MAD与RMSD值,结果见表S3(SI附件,Table S3)与表2和图4。我们可以看到,对于S66全部复合物,APF-D*/6-311++G (2d,p),ωB97X-D/6311++G(3df,3pd)和B3LYP-D3/ Def2-QZVP的RMSD值分别为1.77、1.88、1.50 kJ· mol-1,APF-D*方法计算精度稍优于ωB97X-D,略逊于B3LYP-D3。其中,氢键组的23个复合物,ωB97X-D表现较好,对于17、20、22、23号结合能较大的复合物(大于-75 kJ·mol-1)误差也较小,APF-D*与B3LYP-D3表现相似,结合能较小的1至16号复合物B3LYP表现较好,结合能较大的17至23号复合物APF-D*表现更好一些;对于色散组复合物,APF-D*与B3LYP-D3表现优秀,23个复合物基本都在2.1 kJ·mol-1误差范围,而ωB97X-D对于34至46号结合能小于-21 kJ·mol-1的复合物,绝对误差均在4.2 kJ·mol-1左右,相对误差很大;对于混合类型组,三种方法表现大体相同,B3LYP-D3略好于APF-D*与ωB97X-D。
3.2.2 L7测试集
APF-D*方法对S66测试集表现出令人满意的效果,S66的复合物属于中小尺寸体系,且S66是APF-D*阻尼因子的训练集。对于超出其训练集的大体系APF-D*的表现需要深入测试。我们使用了Hobza研究组的大体系L7测试集进行计算。组成L7测试集的7种复合物分别包含48至112个原子。L7复合物的几何结构是TPSS-D/TZVP方法(未校正BSSE)优化得到,结合能以QCISD(T)/CBS或CCSD(T)/CBS为参考标准。表3中的结合能除了M06-2X和M06-2X-D3外,其余均为经过BSSE校正的结果。
从表4统计结果可以看到,APF-D方法L7的RMSD达到14.17 kJ·mol-1,APF-D*对APF-D计算精度的改善非常明显,其RMSD减少到1.84 kJ· mol-1。ωB97X-D与B3LYP-D3的RMSD分别为5.06和4.01 kJ·mol-1。此外还与L7的其它方法的计算结果进行了比较,M06-2X无色散校正的RMSD高达22.28 kJ·mol-1,经色散校正后M06-2X-D3降到9.07 kJ·mol-1;就MAD指标而言,APF-D*也具有很好的表现,ωB97X-D因GCGC点的离散度大而导致RMSD偏大,而B3LYP-D3方法的离散度较平均,RMSD与MAD的趋势基本一致。APF-D*在几种方法的比较中表现出了最好的计算精度,MAD与RMSD值均小于2.1 kJ·mol-1。
为了测试和比较APF-D*,B3LYP-D3,ωB97XD方法在各自训练集基函数下,获得最好计算精度结果的计算效率,我们在一台Dell R730,配置了Intel Xeon E5-2860双路16核CPU的服务器完成L7测试计算,采用Gaussian 09程序的单节点SMP对称多处理16核并行,即%NProc=16。计算作业的等待时间(Wall Time)也列入表4中。
3.3APF-D*方法势能曲线分析
原始APF-D方法在分子间复合物几何结构优化的测试中具有良好的表现。对APF-D的SAM色散部分做阻尼修正是否会对此产生影响,为此,我们选择了S22测试集中以色散作用为主的15号复合物Adenine-Thymine π…π堆叠碱基对,以传统氢键作用为主的7号复合物Adenine-Thymine Watson-Crick碱基对两个典型的体系来考察APF-D*方法这类应用的可靠性,结合能计算结果都经过BSSE校正。
通过计算S22*5提供的5个距离点的结构结合能对比CCSD(T)/CBS结果可以看到,图5中APFD和改进后的APF-D*均与CCSD(T)/CBS具有相似的势能曲线,势能曲线极小点对应的分子间距离重合得很好。对非平衡结构对应的结合能,曲线整体向下漂移,APF-D方法也是夸大的,对于Adenine-Thymine π…π堆叠碱基对APF-D*的势能曲线与CCSD(T)/CBS几乎重合;而对于Adenine-Thymine Watson-Crick碱基对APF-D*的势能曲线也比APF-D更接近CCSD(T)/CBS。
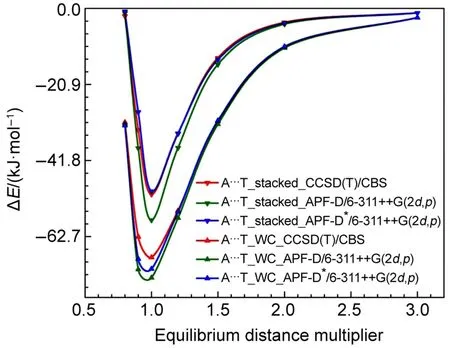
图5 Adenine-Thymine π-π与Adenine-Thymine Watson-Crick复合物在CCSD(T)、APF-D与APF-D*方法中的势能曲线Fig.5 Dissociation energy curves of theAdenine-Thymine complex in the π-π stacked and Watson-Crick conformations using CCSD(T),APF-D,and APF-D*methods
4 结论
APF-D是一种具有较高计算效率的色散校正密度泛函方法,虽然能够较为精确地预测稀有气体及其与小分子复合物的结合能和几何结构,但对于氢键、C―H…π、π…π和C―H…C―H等常见相互作用,结合能计算结果平均偏大13%。导致这种误差的主要原因是APF-D方法SAM模型过度补偿了长程色散能,借助于不同体系色散作用夸大幅度的近似一致性,我们对SAM色散能乘上用统计方法确定的阻力因子,简单、有效地扣除了过度补偿的长程色散能,提出了APF-D*方案。发现APF-D*计算结果对基函数的依赖不大,选择6-311++G(2d,p)基函数是最有效的组合。
计算实践表明,改进后的APF-D*方案除了保持了原APF-D对分子间复合物几何结构优化良好表现外,对S66、L7等分子间相互作用测试集的平均计算结果误差均小于2.1 kJ·mol-1,整体计算精度优于B3LYP-D3和ωB97X-D等被广泛认可的分子间相互作用色散校正密度泛函计算方法。在相同计算精度级别的泛函中,APF-D*具有更高的计算效率,期待它在大体系分子间相互作用研究中能得到广泛应用。
Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn.
References
(1) Lewars,E.G.Computational Chemistry:Introduction to the Theory and Applications of Molecular and Quantum Mechanics; KluwerAcademic Publishers:Massachusetts,2010;pp 5―6.
(2) Zhao,Y.;Truhlar,D.G.J.Chem.Theory Comput.2005,1,415. doi:10.1021/ct049851d
(3) Xu,X.;Goddard,W.A.J.Phys.Chem.A 2004,108,2305. doi:10.1021/jp035869t
(4) Frey,J.A.;Leutwyler,S.Chim.Int.J.Chem.2005,59,511.doi: 10.2533/000942905777676245
(5) Rezac,J.;Hobza,P.Chem.Rev.2016,116,5038.doi:10.1021/ acs.chemrev.5b00526
(6) Grimme,S.;Hansen,A.;Brandenburg,J.G.;Bannwarth,C. Chem.Rev.2016,116(SI),5105.doi:10.1021/acs. chemrev.5b00533.
(7) Grimme,S.;Antony,J.;Ehrlich,S.;Krieg,H.J.Chem.Phys. 2010,132,154104.doi:10.1063/1.3382344
(8) Chai,J.D.;Head-Gordon,M.Phys.Chem.Chem.Phys.2008, 10,6615.doi:10.1039/B810189B
(9) Burns,L.A.;Vázquez-Mayagoitia,A.;Sumpter B.G.;Sherrill, D.J.Chem.Phys.2011,134,95.doi:10.1063/1.3545971.
(10) Austin,A.;Petersson,G.A.;Frisch,M.J.;Dobek,F.J.; Scalmani,G.;Throssell,K.J.Chem.Theory Comput.2012,8, 4989.doi:10.1021/ct300778e
(11) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, Revision D.01;Gaussian Inc.:Wallingford CT,2009.
(12) Mardirossian,N.;Head-Gordon,M.J.Chem.Phys.2015,142, A1113.doi:10.1063/1.4907719
(13) Mardirossian,N.;Head-Gordon,M.J.Chem.Phys.2016,144, 214110.doi:10.1063/1.4952647
(14) Rezac,J.;Riley,K.E.;Hobza,P.J.Chem.Theory Comput. 2011,7,2427.doi:10.1021/ct200294
(15) Sedlak,R.;Janowski,T.;Pitonak,M.;Rezac,J.;Pulay,P.; Hobza,P.J.Chem.Theory Comput.2013,9,3364.doi:10.1021/ ct400036b.
(16) Wang,W.Z.;Sun,T.;Zhang,Y.;Wang,Y.B.J.Chem.Phys. 2015,143,4145.doi:10.1063/1.4931121
(17) Su,P.;Li,H.J.Chem.Phys.2009,131,014102.doi:10.1063/ 1.3159673
(18) Jurecak,P.;Sponer,J.;Cemy,J.;Hobza,P.Phys.Chem.Chem. Phys.2006,8,1985.doi:10.1039/b600027d
(19) Takatani,T.;Hohenstein,E.G.;Malagoli,M.;Marshall,M.S.; Sherrill,C.D.J.Chem.Phys.2010,132,1290.doi:10.1063/ 1.3378024
(20) Podeszwa,R.;Patkowski,K.;Szalewicz,K.Phys.Chem.Chem. Phys.2010,12,5974.doi:10.1039/B926808A
(21) Carter,D.J.;Rohl,A.L.J.Chem.Theory Comput.2012,8,281. doi:10.1021/ct200679b
(22) Dunning,T.H.J.Chem.Phys.1989,90,1007.doi:10.1063/ 1.456153
(23) Kendall,R.A.;Dunning,T.H.;Harrison,R.J.J.Chem.Phys. 1992,96,6796.doi:10.1063/1.462472
(24) Woon,D.E.;Dunning,T.H.J.Chem.Phys.1993,98,1358. doi:10.1063/1.464303
(25) Weigend,F.;Ahlrichs,R.Phys.Chem.Chem.Phys.2005,7, 3297.doi:10.1039/b508541a
(26) Boys,S.F.;Bernardi,F.Mol.Phys.1970,19,553,doi:10.1080/ 00268977000101561
(27) BEGDB online database,http://http://www.begdb.com/ (accessed Mar 1,2016).
An Improvement of the SAM Dispersion Correction in the APF-D Density Functional Method for Studying Intermolecular Interactions
HE Yu1,2WANG Yi-Bo1,2,*
(1Key Laboratory of High Performance Computational Chemistry,Guiyang 550025,P.R.China;2Network and Information Center of Guizhou University,Guiyang 550025,P.R.China)
Austin-Petersson-Frisch(APF)is a new hybrid density functional method that combines B3PW91 and PBE0.APF-D provides an additional empirical dispersion correction method based on a spherical atom model(SAM),which is different from the Grimme′s empirical dispersion correction method.APF-D accurately describes the binding energy and the potential energy surfaces of complexes of noble gas atoms and small hydrocarbon dimers.However,APF-D is not accepted as a standard method to study intermolecular interactions because the results often show a large deviation from the normal range when using the APF-D method to calculate the binding energy of hydrogen bonded complexes,C―H…π and π…π interactions.Our research identified that such a deviation arises from some long-range dispersion that has been double counted by the APF function and the SAM dispersion correction.Therefore,we propose an improved APF-D method,termed APF-D*.By taking advantage of ζ,which is independent of SAM dispersion,we were able to solve effectively the problem of excessive dispersion compensation in APF-D.By comparing the results from S66 and L7 benchmark sets,we find that APF-D*greatly improved the precision of calculations over the traditional APF-D method.The overall accuracy of APF-D*was found to be comparable to or better than current leading DFT methods,such as B3LYP-D3 and ωB97X-D.However,both B3LYP-D3 and ωB97X-D have a much larger computational cost thanAPF-D*.We believe that APF-D*is a better method to calculate of the intermolecular energy of large molecules.
O641
10.3866/PKU.WHXB201609132
Received:July 11,2016;Revised:September 12,2016;Published on Web:September 13,2016.
*Corresponding author.Email:ybw@gzu.edu.cn;Tel:+86-851-88292009.
The project was supported by the Natural Science Foundation of Guizhou Province,China(20082116).
贵州省自然科学基金(20082116)资助项目
