3C-like蛋白酶抑制剂的构效关系、分子对接和分子动力学
2016-12-29付新梅蒋思宇王景辉张述伟
林 峰 付新梅 王 超 蒋思宇 王景辉张述伟 杨 凌 李 燕,*
(1大连理工大学化工学院,辽宁大连116024;2大连理工大学精细化工国家重点实验室,辽宁大连116024;3中国科学院大连化学物理研究所,药物资源开发研究组,辽宁大连116023)
3C-like蛋白酶抑制剂的构效关系、分子对接和分子动力学
林 峰1付新梅2,*王 超1蒋思宇1王景辉1张述伟1杨 凌3李 燕1,*
(1大连理工大学化工学院,辽宁大连116024;2大连理工大学精细化工国家重点实验室,辽宁大连116024;3中国科学院大连化学物理研究所,药物资源开发研究组,辽宁大连116023)
3C-like蛋白酶是中东呼吸综合征冠状病毒(MERS-CoV)等其它冠状病毒的繁殖过程中极为重要的蛋白酶。它已成为人类在抗冠状病毒领域中的研究热点。本文基于计算生物学方法对与MERS-CoV同属的蝙蝠冠状病毒HKU4(HKU4-CoV)的43个肽类3C-like蛋白酶抑制剂分子,建立三维定量构效关系(3D-QSAR)模型。在基于配体叠合的基础上,发现比较分子相似性指数分析法(CoMSIA)中的四个场组合(位阻场、静电场、氢键供体场与氢键受体场)为最优的模型(Q2=0.522,R2ncv=0.996,R2pre=0.904;Q2:交叉验证相关系数,R2ncv:非交叉验证相关系数,R2pre:验证集分子的预测值相关系数),并借助该模型通过分子对接(docking)与分子动力学(MD)方法阐明了配受体结合作用。实验结果表明:(1)基于最优的CoMSIA模型基础上的三维等势图形象地说明了分子基团的位阻作用、静电作用、氢键供体与氢键受体作用对分子生物活性的影响;(2)分子对接研究结果显示了疏水性以及结晶水、氨基酸His166和Glu169在配体和受体结合过程中产生重要作用;(3)分子动力学模拟进一步验证了分子对接模型的可靠性,并发现了两个新的关键氨基酸Ser24与Gln192,它们与配体产生了两个较强的氢键。此外,根据这些结果,一些新的具有潜在抑制活性的肽类化合物作为3C-like蛋白酶抑制剂被获得。以上结果能够帮助深入了解3C-like蛋白酶与肽类抑制剂的作用机理,并且能够为今后的抗MERS-CoV药物设计提供有价值的参考。
中东呼吸综合征冠状病毒;3C-like蛋白酶;肽类抑制剂;三维定量构效关系;分子对接;分子动力学
1 引言
冠状病毒(CoV)是一类分布广泛的,具有重大威胁的病原体1,2。该种病毒能引起人及哺乳类动物疾病,且被感染的人或动物可能会成为呼吸道、肠道、肝脏、神经系统疾病的携带者。最近出现的中东呼吸综合征冠状病毒(MERS-CoV)就是其中能感染人类并传播的冠状病毒之一3。该病毒于2012年9月在中东地区首次被鉴定出来,随后逐渐扩散,在很多国家都报道了感染病例。MERS-CoV能导致非常严重的呼吸道病症,致死率高。截至2015年1月30日,共有956例确诊病例,死亡351例,死亡率高达36.7%,远高于严重急性呼吸综合征冠状病毒(SARS-CoV),引发了全球关于MERS大流行潜在可能性的恐慌4。因此,MERS-CoV已经成为一种全球范围内严重威胁人类生命安全的新型冠状病毒。
冠状病毒被国际病毒分类委员会(ICTV)分为四个属,即α、β、γ和δ冠状病毒属。其中,β冠状病毒属有四个亚群(A-D),通过将MERS-CoV和其他β冠状病毒基因序列对比发现,MERS-CoV与SARS-CoV中较保守的核酸复制酶序列只有小于50%的氨基酸相同,而与从蝙蝠体内分离到的蝙蝠冠状病毒HKU4(HKU4-CoV)和HKU5 (HKU5-CoV)亲缘关系很近,相似度分别约为75%和76.7%5-7。因此,ICTV在β冠状病毒属C亚群中增加MERS-CoV为一个新种,它也是该亚群中第一种能够感染人类的β冠状病毒。研究表明,MERS-CoV不仅可存在于蝙蝠体内,其也被发现能生存在卡塔尔单峰骆驼体内,而且它们的辅助蛋白在体外能够抑制人类抗病毒的信号路径3,8-10。除此之外,Wang等11通过研究认为MERS-CoV的起源是HKU4-CoV或HKU5-CoV的变异,其中更多研究表明,其更倾向是来自HKU4-CoV的变异,表明从蝙蝠到人类的一种人畜共患的转变。
3C-like蛋白酶(3CLpro)是病毒繁殖的蛋白水解过程中,两个非常重要的病毒半胱氨酸蛋白酶之一12-14。3C-like蛋白酶在拥有典型的Leu-Gln (Ser、Ala、Gly)氨基酸序列的11个保留位点上将复制酶多聚蛋白断裂,其中它们的P1位置与P2位置分别拥有完整的Gln氨基酸残基和脂肪族氨基酸残基15,16。迄今为止,没有疫苗和抗病毒药物能够真正有效预防冠状病毒感染人类或治疗已患病的人类。因此,3C-like蛋白酶对于冠状病毒生命周期起着至关重要的作用,使其成为一个抗MERSCoV药物的发展很有吸引力的目标,基于3C-like蛋白酶抑制剂的研究具有广阔的前景17-19。
随着计算机科技的高速发展,三维定量构效关系(3D-QSAR)作为一个十分有效、经济的方法,已被广泛应用于各个领域的结构与活性、结构与性质关系的研究,来帮助设计新的药物20,21。3DQSAR包括比较分子力场分析(CoMFA)和在其基础之上发展起来的较新颖的比较分子相似性指数分析(CoMSIA)等众多方法。这些方法认为,配体与受体之间的相互作用取决于化合物周围分子力场的差异,以定量化的分子力场参数作为变量,对药物活性进行回归分析便可反映出药物小分子与生物大分子之间的作用模式,进而有选择地设计新药。
目前MERS-CoV的3C-like蛋白酶抑制剂还处于研究阶段,但已有文献证明与MERS-CoV同属的SARS-CoV的3C-like蛋白酶抑制剂也同样能良好地抑制MERS-CoV的繁殖22-25。本文通过上述计算方法,对新合成的与MERS-CoV更接近的HKU4-CoV的43个肽类3C-like蛋白酶抑制剂分子进行了研究,建立了3D-QSAR模型,并探讨了此类抑制剂周围的位阻场、静电场、氢键供体场和氢键受体场对生物活性的影响。此外,我们还利用分子对接方法来验证3D-QSAR模型的准确性和稳定性,之后采用分子动力学方法对分子对接的结果进行补充。它们能帮助我们更好地预测抑制剂小分子与靶点蛋白大分子之间可能的结合构象,进而预测小分子与蛋白的结合力和其生物活性。我们希望获得的分子构效关系以及配受体结合作用的结论能够延伸到为今后抗MERS-CoV药物(3C-like蛋白酶抑制剂)的设计提供理论指导。
2 实验部分
2.1数据库及生理活性
本文模型的建立及分析采用的是Tripos公司的SYBYL 6.9设计软件包。文中所研究具有抑制3C-like蛋白酶作用的肽类化合物结构和活性数据均来自于文献26。这些分子的抑制活性范围IC50较宽,将其值转化pIC50(-logIC50)值作为定量构效关系分析中的因变量,使数值均匀地分布在4.25-6.50之间。整个数据集以近似3:1的比例,被分为两个部分。其中,33个分子结构和活性作为训练集,用于模型的构建;而其余10个分子则加入到验证集中,用于测试该模型的准确性。训练集和验证集的选取应遵循随机挑选的原则,在尽可能满足更好的结构差异性与多样性的同时,使训练集与验证集的分子比较均匀地分散在整个数据库范围中。我们将所有的分子结构及其对应的生理活性pIC50实验值列于表S1(Supporting Information)中。其中,表S1还包含基于配体和受体的CoMFA与CoMSIA模型得到的pIC50预测值,表S1粗体字显示的部分是具有最佳预测性能的模型的pIC50预测值。
2.2构象优化及分子叠合
在建立3D-QSAR模型中,分子叠合是十分重要的环节,其直接决定了模型的优劣,而在分子叠合前需要进行构象优化27。分子三维构象优化的步骤如下:
(1)采用Chemoffice软件构建化合物分子的二维和三维结构;
(2)导入到SYBYL中进行能量优化,每个分子上加载Gasteiger-Hückel电荷28;
(3)利用Tripos力场29作为能量最小化和构象搜索的共轭方法,且收敛条件设置为0.00419 kJ· mol-1;
(4)采用Powell能量梯度获得稳定构象,且最大优化次数设置为1000次,梯度设置为2.095 kJ· mol-1·nm-1。
在整个数据集中,我们选择活性最大的1号分子进行叠合,应用SYBYL软件,在1号分子上划定所有分子的分子骨架,用紫色线显示,如图1a所示。在研究中,使用了三种不同的叠合方式:叠合I是基于配体的叠合,得到叠合图为图1b;叠合II是基于受体完成的,将所有分子都对接于受体中,得到对接结果最高打分的构象进行叠合,分子叠合图为图1c;叠合III也是一种基于受体的叠合方法,其中所有的分子构象均来自于叠合II,随后经历叠合I的叠合过程,并得到叠合图为图1d。
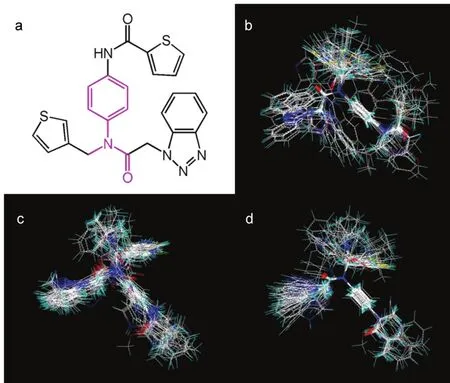
图1 模板分子结构及三种分子叠合图Fig.1 Structure of template molecule and three kinds of molecular alignment
2.3CoMFA及CoMSIA研究
CoMFA及CoMSIA都是应用最广泛的合理药物设计方法。CoMFA基于的假设是配体与受体的相关作用是非共价性的相互作用,利用分子力学力场(位阻场和静电场)可以较好地解释它们的相关作用30。相比之下,CoMSIA是以CoMFA为基础,并对其进行了补充和优化。CoMSIA将CoMFA中的位阻场和静电场进一步细分为五个场,即多引入了疏水、氢键供体和氢键受体场,所以更细致地描述了配体与受体间的相互作用。此外,CoMSIA改变了探针原子与配体之间作用能的计算公式,非CoMFA传统的库仑(Coulomb)和范德华(Lennard-Jones)函数,而采用了更加平滑的高斯(Gaussian)函数来计算分子场的数据,从而有效避免了原子位置发生异常和配体周围不同探针位点上势能的差异31。而且在CoMSIA中,分子力场能量于不同的探针位点间衰减很快,致使计算过程自动收敛,无需再设定能量截断值。
在本文的研究中,我们采用了CoMFA和CoMSIA两种定量构效关系方法,用来间接地反映了化合物分子与受体相互作用过程中两者之间的非键相互作用特征,并预测良好的三维定量构效关系模型。分子叠合后,分子空间取向基本一致,然后用一个粒子探针在分子周围的空间中游走,计算粒子探针与分子之间的相互作用,并记录下在空间不同坐标中它们相互作用的能量值,进而获得分子力场数据。在CoMFA建模方法中,用sp3杂化的C+为探针,探针所带电荷为+1.0e,半径为0.152 nm,默认步长为0.2 nm,对整个三维网格进行搜索,且位阻场和静电场效应能的阈值分别设定为默认值209.29 kJ·mol-1,同时其它参数均为默认值。对于CoMSIA建模中,用到的三维网格和CoMFA相同,以sp3杂化的C+为探针,电荷为+1.0e,半径为0.100 nm,疏水性为+1.0,氢键供体和受体强度均为+1.0。此外,构建这两种的QSAR模型时,衰减因子均设定为默认值0.3。
2.4PLS分析与验证
本文采用偏最小二乘回归分析(PLS)来建立CoMFA/CoMSIA描述符和抑制剂pIC50值之间的关系。在PLS的建模中,为了避免过度拟合问题,必须要对其中每个连续组分的重要性进行严格测试,然后测试会在组分不重要时停止。交叉验证是一种能测试这种重要性的实用且可靠的方法32。我们运用的交叉验证方法是抽一法(leave-oneout),它是依次从训练集中抽取一个样本,余下的n-1个样本作为训练集构建模型,并用该模型预测抽取出的样本,直到每个分子都被取出预测一次,最终可以得到最佳组分数(OPN),与能够衡量所建模型的内部预测能力的重要统计指标,交叉验证相关系数(Q2)。而利用所有训练集的分子结合OPN,通过非交叉验证分析计算可以得出非交叉验证相关系数(R2ncv)、统计值(F)、训练集的估计标准误差(SEE)和验证集的最小标准预测偏差(SEP)33。此外,根据预测验证集的活性,我们还可以得到能够评估模型的外部预测能力的重要参数,验证集分子的预测值相关系数(R2pre)。因此采用PLS能在避免过拟合现象和消除大多数可变因素的同时,建立大量的QSAR方程34,35。最后,我们将CoMFA/CoMSIA结果以等势图的形式直观地表示出来,并进行下一步的分析。
2.5分子对接
为揭示肽类抑制剂小分子和蛋白大分子之间的结合机制,以及确定抑制剂的生物活性构型,我们从Protein Data Bank数据库36中选择蛋白靶点的三维晶体结构PDB ID:4YOI26,基于遗传算法的GOLD 5.0版本软件37,进行分子对接研究。分子对接是利用配体小分子与受体蛋白大分子的完整结构信息,计算把配体放在受体活性位点的位置,然后按照能量匹配、几何匹配以及化学环境匹配的原则来评价抑制剂和蛋白的结合能力,并预测它们之间最佳的结合模式38。在实际过程中,不仅配体小分子具有柔性易产生多种构象,而且受体大分子也是柔性且构象可发生变化。在两者分子对接过程中,它们的构象会变化以找到一个能达成局部稳定的契合点。因此,研究人员可以通过分子对接确定柔性的配体与受体蛋白的结合位置和空间取向,以此来研究药物的作用机制并指导新药物的设计。
分子对接之前,先对蛋白质模型进行修改,只留下单链,并在蛋白质结构中加入极性氢,且删除原有的配体与杂原子。我们在GOLD中将受体大分子中每一个原子坐标1 nm空间内的氨基酸残基所组成的空间范围设为可能与配体小分子的结合位点。我们将43个分子分别对接到潜在的结合位点中,每次对接都会产生5个左右可能的构象。所有构象都会经过GOLD评估,对其适当性进行打分。打分的分值计算考虑到了配体分子内氢键和拉力的作用,以及配体与受体之间相互的氢键和范德华力的贡献39。在分子对接过程中,打分函数采用GoldScore算法,其他参数采用软件中的默认值。
2.6分子动力学
为了进一步检验分子对接研究中结果的稳定性,以及构建一个能够表现分子所在真实环境的蛋白质模型,即需要考虑蛋白质大分子的动态灵活性,我们应用GROMACS软件包中的GROMOS96力场40,41对分子对接得到的复合物开展了详尽的分子动力学模拟。分子动力学模拟是研究生物大分子体系的常用计算方法。该法根据牛顿经典力学原理,模拟分子体系的运动状态,计算在相空间中所有原子的运动轨迹,接着在从各种状态的分子体系构成的系综中,选取样本计算构型积分,且以此为基础,进行计算该体系的热力学量等其他宏观性质。
进行动力学模拟之前,需要对模拟体系进行一系列预处理。我们在PRODRG 2.542中获得配体-蛋白质复合物分子的拓扑文件,随后将初始复合物放置在一个边长为10 nm的周期性立方体晶格体中,其中保持晶格上每一点与蛋白质的距离至少是1 nm41。为了保证体系的电中性,蛋白质复合物和水中所有原子均被随机地放置于晶格盒子中,并用简单点电荷水来填充模拟体系的其余部分43。详细的动力学模拟步骤如下:首先为了避免分子间高能碰撞的可能性,对整个系统进行2500步最快下降法操作,令其能量逐步最小化,再进行2500步共轭梯度优化。接着,当整个系统的温度和压力上升到300 K和101.325 kPa且处于每纳米8.38 kJ·mol-1稳定力时,对常温常压NPT系综进行50 ps恒压力与500 ps密度平衡操作。最后,在周期性边界条件下,设定积分步长为0.002 ps,在保持恒温的条件下进行50 ns动力学模拟。在整个模拟过程中,我们分别采用了SHAKE算法44与PME方法45来固定含碳氢键和处理库仑作用。
3 结果与讨论
3.1 CoMFA与CoMSIA模拟统计量分析
在之前的研究工作中,我们分别运用了基于配体和基于受体的三种叠合方式对数据集中43个肽类分子进行叠合,生成了六个不同的叠合模型,并对CoMFA两种力场(位阻场与静电场)和CoMSIA五种力场(位阻场、静电场、疏水场、氢键供体场和氢键受体场)的所有3种与31种排列组合进行了计算。为了提高模型的可对比性,所有的CoMFA和CoMSIA模型在建模过程中都使用相同的训练集(33个分子)和验证集(10个分子)。通过对比模型结果,得出基于配体得到的模型最优,因此我们主要分析基于配体的模型结果。这些模型的统计参数列于表1中。
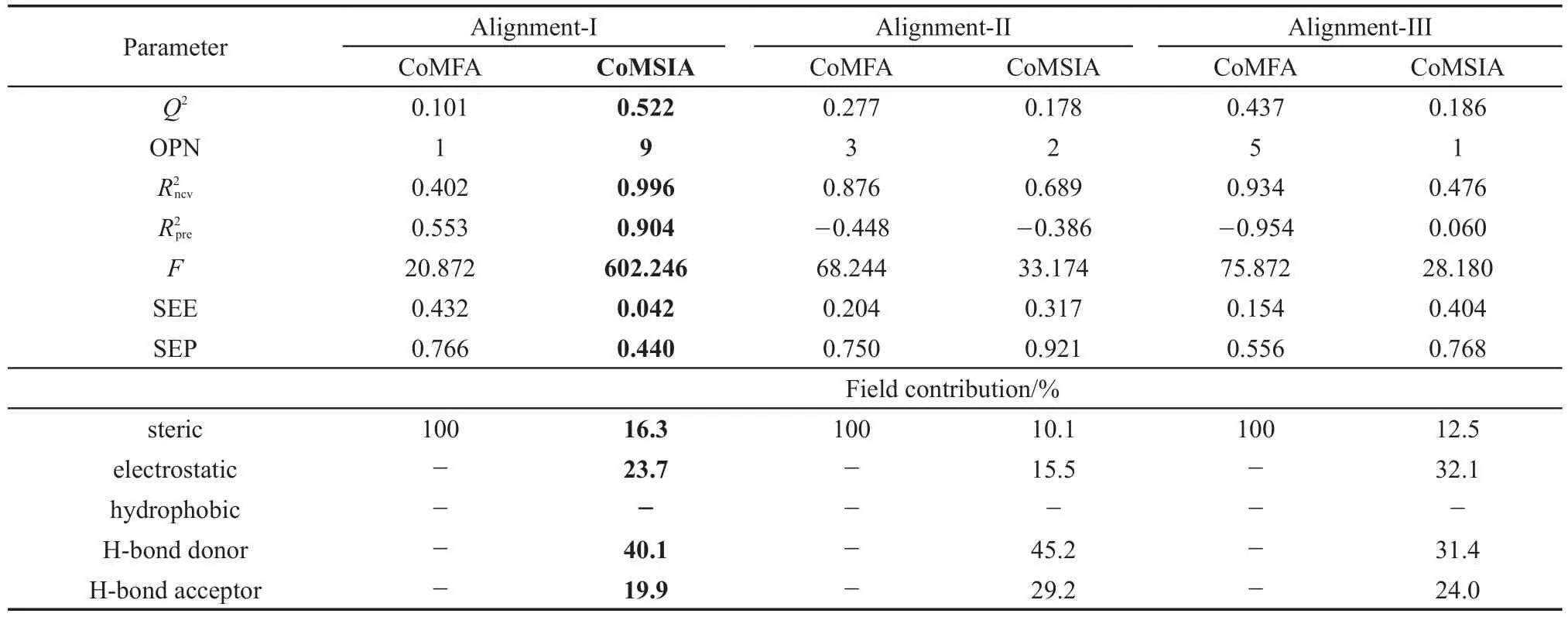
表13D-QSAR结果汇总Table 1 Summary of 3D-QSAR results
对比得到了一个具有最佳预测性能的模型结果(表1中黑体字显示),并用PLS法和抽一法对其进行评估,获得包括用来检验所建模型是否有效的3个参数(Q2、OPN与R2ncv)和用来检验模型预测能力的验证集R2pre。此外,还包括一些其他的统计参数,如模型标准误差,即F值、训练集的SEE和验证集的SEP,以及各个力场对模型的贡献率。
从统计意义上来说,一个良好的3D-QSAR模型,前提必须满足四个重要条件:Q2>0.5且,并有较低的SEE、较大的F值,才能很好地预示CoMFA和CoMSIA模型46。因此我们通过建模分析,最终以组合了拥有最高的的位阻场、静电场、氢键供体场和氢键受体场的CoMSIA模型为最优模型。其Q2=0.522、OPN= 9、R2ncv=0.996、F=602.246和SEE=0.042,可以看出这个模型的误差较小且可靠性较高。当被验证集验证时,所得模型R2pre=0.904与SEP=0.440,展示出其良好的预测能力。所有这些数据结果都说明了我们所建立的基于配体叠合的CoMSIA模型是一个可靠的具有很好预测能力的模型。
同时,图2中显示了在最优的CoMSIA模型中,所有训练集分子和验证集分子的预测pIC50值和实际pIC50值的散点图。其中训练集分子以方块代表,而验证集分子以三角表示。从图中可以看出,整个数据集都是密集地分布在回归线的两侧,而且抑制剂于模型中预测的pIC50值与实验中测得的pIC50值的差值较小。这表明训练集和验证集的密集度和误差都很相似,未出现过拟合时训练集分布均匀,而验证集误差较大(离回归线很远)的问题,且表明预测值和实际值的相关性良好,得到了比较可靠的构效关系模型47,48。
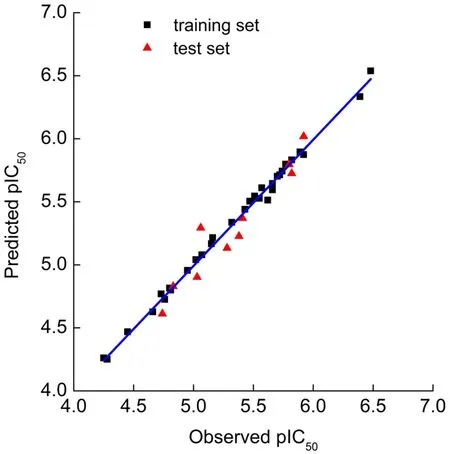
图2 基于配体的CoMSIA模型的预测与实际pIC50值的关联图Fig.2 Ligand-based correlation plot of the predicted versus the actual pIC50values based on the CoMSIAmodel
3.23D-QSAR等势图结果与分析
为进一步形象化和具体化得到的CoMSIA模型中包含的信息和内容,我们分析CoMSIA模型中四个力场的等势图。这些等势图能够直观地反映抑制剂小分子的生物活性与其化学结构上的相互联系,从而探究出对药物分子活性有关的重要因素,进而在实际药物设计中提供指导49。在绘制等势图过程中,为了能够更好地分析各种等势图的结果,一致将活性最高的分子,1号分子(pIC50= 6.481)选为模板分子,叠放于四张等势图(图3)中,以便于对比。在该1号分子的三维结构(图4)中,R1、R2、R3和R4取代基是数据集内其他分子结构与1号分子的差别所在。此外,在该CoMSIA模型四个力场的等势图中,积极影响区域和消极影响区域贡献比率分别设置为默认值80%和20%。
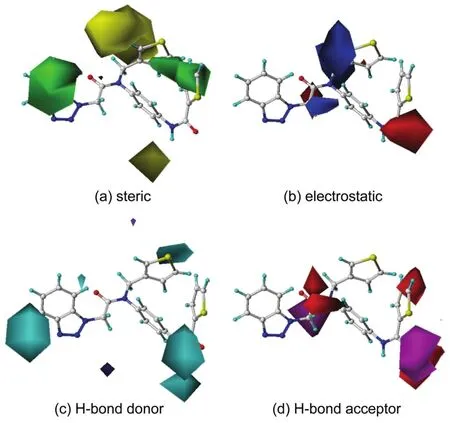
图3 结合1号分子的CoMSIA模型等势图Fig.3 CoMSIAcontour maps in combination with compound 1
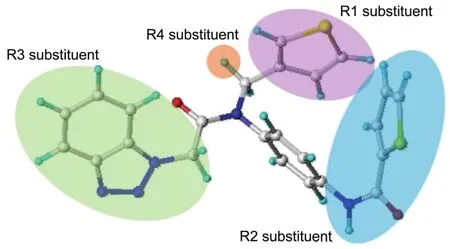
图4 1号分子结构Fig.4 Structure of compound 1
图3(a)为基于1号分子CoMSIA位阻场的等势图。其中,绿色和黄色云团分别表示位阻基团对分子活性的有利和有害区域。由图可看出,R3取代基远离分子骨架的部分和远处R1与R2取代基之间有很大的绿色区,说明R3取代基远位以及R1与R2取代基相近部分存在的较大基团有益于提高分子活性,这一点从数据集中得到了充分的验证。例如,24号分子(pIC50=5.658)与34号分子(pIC50= 4.954)对比,以及1号分子(pIC50=6.481)与13号分子(pIC50=5.509)对比,它们的差别只在对应位置存在较大基团,使位阻变大,有益于提高抑制活性。此外,R2取代基靠近分子骨架苯环的部分和R4取代基周围存在两块黄色云团,显示了位阻较大基团在这片区域对分子活性起抑制作用。例如2号分子(pIC50=6.387)和28号分子(pIC50=5.377)的R4取代基中,H原子的位阻明显小于叔丁基的位阻,使得28号分子较2号分子活性低很多。
图3(b)为基于1号分子CoMSIA静电场的等势图。其中,蓝色云团代表正电性积极影响区域;而红色云团表示负电性积极影响区域。图中R4取代基临近分子骨架的部分有大片蓝色区域,说明此处正电性取代基的存在对提高抑制活性有利,如在此处带有正电性的叔丁基基团的分子活性较大。而图中R2取代基临近分子骨架的部分有大片红色区域,另外还有小片红色区域在R3取代基靠近分子骨架氧原子的部分,说明负电性取代基在此处有利。图可见,R2取代基临近分子骨架部分和R1与R3取代基远离分子骨架部分附近有较大的青色区域,说明该区域有氢键供体作用的取代基对提高分子活性有利。例如,最高活性的1号分子在R2取代基处有N―取代基(其上拥有氢原子作为氢键供体),其抑制活性远远高于其它无氢键供体基团或N―取代基(其上未拥有氢原子)的类似物分子,即是一很好的佐证。总的来说此处氢键供体基团能提高抑制剂活性。同时,R3与R4取代基的远位有两个极小紫色云团,其对分子活性具有一定消极影响,但从大趋势来看,基本可以忽略。
图3(d)为基于1号分子CoMSIA氢键受体场的等势图。其中,紫色云团代表更依赖氢键受体区域,氢键受体在此处更益于生物活性,而红色云团则相反。从图可见,紫色与红色云团基本都同时出现在R2与R3取代基靠近分子骨架部分,但它们都处于非同侧,此外还有一块红色云团在R2取代基离骨架的远位。这使分子具有一定的敏感性,说明R2与R3取代基的空间旋转角度不同,导致其受氢键受体场的影响有巨大差异。当R2与R3取代基能作为氢键的接收基团时,且接收基团都恰好旋转到空间中氢键受体有益活性区域时,积极作用达到最大;反之,消极作用最大。例如,在基于9号分子(pIC50=5.699)的CoMSIA氢键受体场的等势图(图5)中,可以看到,9号分子的R2取代基上具有一个S原子和两个N原子可作为氢键受体,R2取代基的角度已基本达到使它们靠近紫色积极云团而远离红色云团;同样,该现象也可从这个分子的R3取代基的三个N原子上看到。氢键受体场对9号分子的积极作用最大,这可能是证明该分子在数据集中具有较高活性的原因之一。
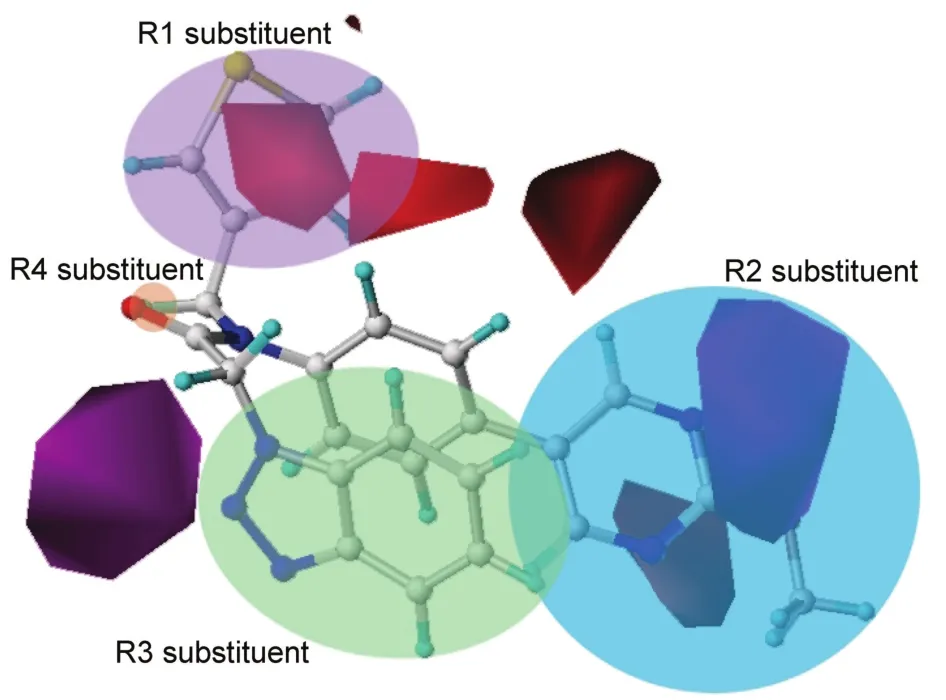
图5 结合9号分子的CoMSIA模型氢键受体轮廓图Fig.5 CoMSIAH-bond acceptor contour map in combination with compound 9
综合以上3D-QSAR的研究结果,我们分析了影响分子抑制活性的各基团作用域(图6)。我们能够得出概括性的关于肽类分子的结构性优化方面的结论:
(1)对于R1取代基,远离分子骨架的部分(靠近R2取代基的周围),如图6红色区域内,大位阻基团和氢键供体基团有利于分子活性的提高;
(2)对于R2取代基,临近分子骨架的部分,如图6深蓝色区域内,负电性基团、氢键供体基团和氢键受体基团有利于分子活性的提高;此外,靠近R1取代基的周围,如图6浅蓝色区域内,大位阻基团有利于分子活性的提高;
(3)对于R3取代基,临近分子骨架的部分,如图6深绿色区域内,负电性基团和氢键受体基团有利于分子活性的提高;此外,远离分子骨架的部分,如图6浅绿色区域内,大位阻基团和氢键供体基团有利于分子活性的提高;
(4)对于R4取代基,临近分子骨架的部分,如图6黄色区域内,正电性取代基有利于分子活性的提高。
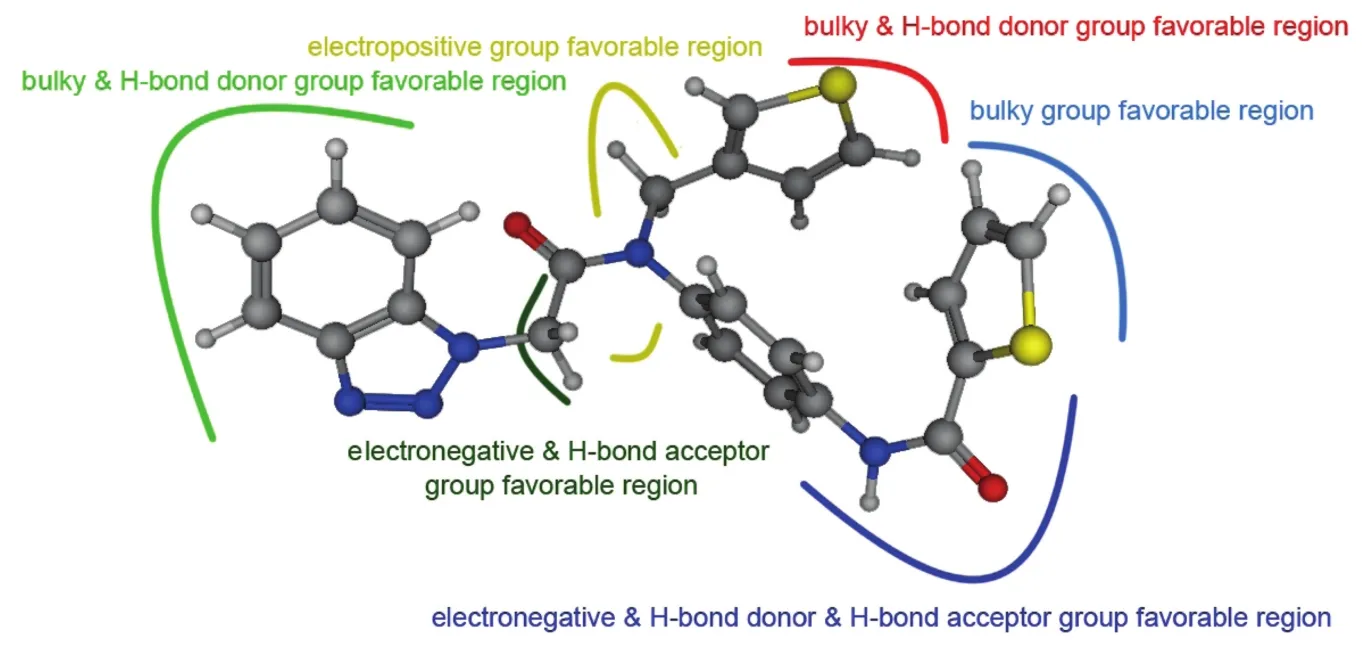
图6 分子抑制作用的交互特性Fig.6 Interaction features of compound impacting the inhibitory effect color online
3.3分子对接研究
分子对接是一种用于验证3D-QSAR模型准确性和稳定性的有效工具。它能帮助我们更好地预测候选药物小分子与靶点蛋白之间可能的结合构象,进而预测小分子的结合力和生物活性。在本文的研究中,为了阐明所选肽类3C-like蛋白酶抑制剂分子能否调节靶点蛋白,同时为了进一步形象化地展现它们之间的作用机制及结合构象,我们对整个数据集中所有43个肽类分子都进行了对接研究。
为了之后能与John等26已做的X射线晶体实验结果作对比分析,我们采用相同的条件(存在结晶水)的受体和配体分子做了分子对接。另外,我们也对比了不存在结晶水的受体和配体分子的对接结果。在它们的对接结果中,获得得分最高的合理性打分,其平均分分别为81.519分与78.922分。从打分结果来看,与存在结晶水的对接结果相比,没有结晶水的对接打分变低了2.597分,受体存在结晶水的对接效果确实比较好,验证了John等26实验条件的合理性。通过对存在结晶水的对接结果分析得到了结晶水HOH523与配体分子产生的氢键距离图(图7),其中它们的氢键距离范围从0.234至0.334 nm。一般来说,形成氢键的几何标准是距离小于0.35 nm,且它们之间所连成的角度必须大于120°,此外0.32-0.40 nm之间的距离可以称为是弱氢键50。因此,HOH523能与大部分的配体(30/43)产生较强的氢键作用,表明了由于它们的氢键存在使配体更加稳定的结合于空腔中,进而影响到整体打分。最终,我们选择1号配体分子与存在结晶水的受体大分子进行分子对接获得的分数最高的、最合理的结果来进行接下来的分析。
图8为1号分子和原来蛋白质晶体中结合的分子互相叠合的结果,它们分别呈橙色和绿色。从图中可明显看出,两个分子在受体中的位置极其相似,这有力地证明了分子对接结果的准确性,能够再现3C-like蛋白酶与其抑制剂分子间的实验结合空腔。
在配体药物分子与受体蛋白相结合时,其主要作用一般是与其周围的氨基酸形成的疏水、氢键等相关作用。图9展示了最高活性的1号分子在其周围0.45 nm内的蛋白质空腔中的氨基酸残基的对接作用模式,包含的氨基酸从图中可以看出,抑制剂分子稳定在蛋白质空腔内,形成一个稳定的空间区域。从图中配体分子周围的氨基酸类型判断,该分子结合的空腔有两个疏水位点(文中标为S1和S2)。S1位点由氨基酸Ser24、Met25、ILe42、Pro45、Ala46以及Leu49等氨基酸构成。1号抑制剂分子的R2取代基部分正结合于此。S1位点中的氨基酸大部分都具有脂肪族氨基酸的特性,它们共同促进该疏水位点的形成。同时,1号抑制剂分子的R3取代基部分也被一个疏水性的S2位点所固定。这些氨基酸包括Leu27、Phe143、Leu144、Cys145、Met165、His166、Met168以及Glu169等,它们也有较强的脂肪特性使得配体分子能够紧密固定在这个空腔中。
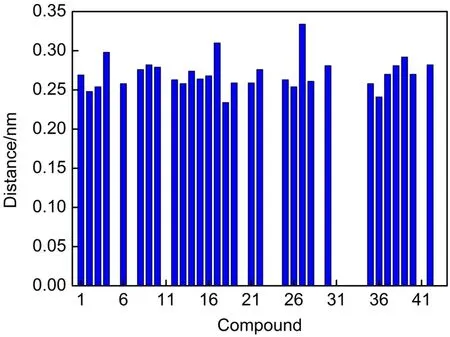
图7 结晶水HOH523与配体分子的氢键距离Fig.7 H-bond lengths between HOH523 and ligand molecules
此外,以上对接结果图显示,配体还通过不同方向的四个氢键紧密地结合在受体空腔内。四个氢键分别为:
(1)配体分子R2取代基上的氮原子与结晶水HOH523氧原子形成的氢键(N―H…O,0.269 nm);
(2)配体分子R3取代基上的远离骨架部分的氮原子与氨基酸His166侧链上氮原子形成的氢键(N…H―N,0.290 nm);
(3)配体分子骨架上的氧原子与氨基酸Glu169骨架上氮原子形成的弱氢键(O…H―N,0.359 nm);
(4)配体分子R3取代基上的靠近骨架部分的氮原子与氨基酸Glu169骨架上氮原子形成的弱氢键(N…H―N,0.352 nm)。
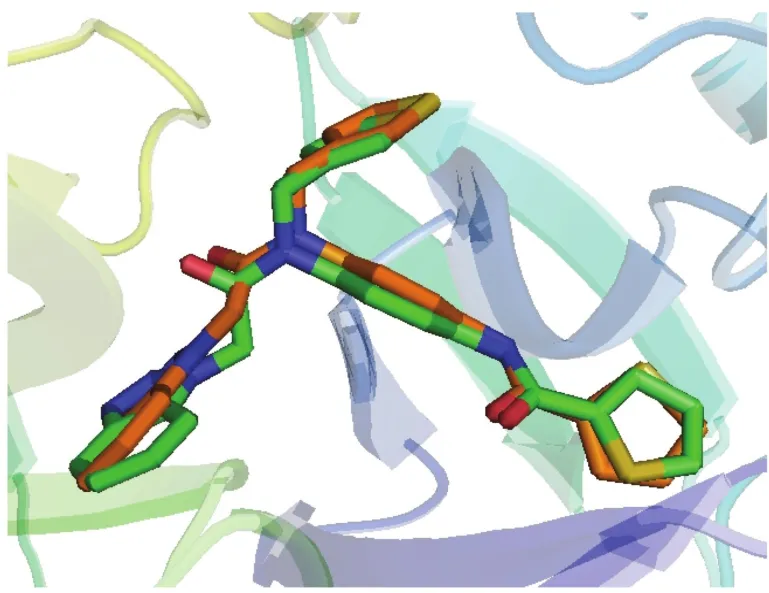
图8 对接在3C-like蛋白酶中的1号分子的结合模式(橙色)和原1号分子(绿色)Fig.8 Binding mode of compound 1(orange)and the original molecule(green)docked in 3C-like protease

图9 对接在3C-like蛋白酶中的抑制剂分子的结合模式Fig.9 Binding mode of inhibitor compound docked in 3C-like protease
与之前的CoMSIA等势图的结果结合分析,由CoMSIA氢键供体场的等势图(图3(c))表明,在氢键供体场的积极作用域内,R2取代基上的氮原子作为氢键供体和结晶水HOH523形成氢键;而由CoMSIA氢键受体场的等势图(图3(d))表明,在氢键受体场的积极作用域内,骨架上的氧原子和R3取代基上的靠近骨架部分的氮原子都作为氢键受体和氨基酸Glu169形成氢键。并且,CoMSIA静电场的等势图(图3(b))显示,R4与R3取代基周围分别存在有正电与负电云团,从对接结果中得知,在这两个位置分别看到了正电性的氨基酸His41与Lys191和负电性的氨基酸Glu169。此外,由于Phe143、His166及His175等氨基酸中相对较大的苯环或杂环结构的存在,也使得在R3取代基附近引入大型取代基会产生较强的位阻效应,这与CoMSIA位阻场的等势图(图3(a))中大块的绿色云图所代表的含义也是十分一致的。这些区域中氢键的形成以及正负电性与含苯环或杂环结构氨基酸的存在有利于提高生物的抑制活性。分子对接的结果与3D-QSAR等势图结果非常吻合,它们互为验证和补充,充分说明了3D-QSAR模型的合理性。
同时,我们也将分子对接的结果与John等26已做的X射线晶体实验结果(PDB ID:4YOI、4YOG与4YOJ)进行了对比。其中,氢键的对比结果如表2所示。从表中可以看出对接结果所得的四个氢键同样在该实验结果中出现,且它们的差别仅在作用力大小的不同,充分说明对接结果的正确性和稳定性。此外,他们实验结果中的关键氨基酸,如氨基酸Met25、His41、Cys44、Ala46、Tyr54、Cys148、His166、Glu169和Gln192都出现在了分子对接的结合空腔中,也说明了分子对接是合理的,表明对接结果有很强的可靠性。
3.4分子动力学研究
对于配体分子结合机制的探明是一个重要的研究步骤,因此我们需要获得肽类抑制剂小分子和3C-like蛋白酶大分子复合物更加真实的结合机制。分子动力学模拟正是能够实现这种优化的方法之一51。分子动力学模拟可以有效地表达分子体系的状态和行为随时间的变化情况。相比属于半柔性模拟方法的分子对接研究,分子动力学方法属于动态模拟的过程,能够高效迅速地搜索出配体分子的低能量构象,并对其构象变化的轨迹进行追踪和表现。实验中,我们采用了水环境下分子动力学模拟的方法来估计所得配体分子的结合亲和性,并进一步评估分子对接中结合机制模型的可靠性。
3.4.11号分子的分子动力学研究
我们以分子对接中的配体-蛋白质复合物作为初始结构,对1号分子进行了50 ns的分子动力学研究,其发生构象改变的动力学图像如图10所示。其中,我们采取了通过对最初构象的监测从而对结构差异性进行几何学测量的方法,均方根偏差(RMSD),来进一步地确保取样方法的合理性与复合物大分子的动力学稳定性。图10a分别展现了1号复合物(黑色)、蛋白(红色)与1号配体(蓝色)的分子动力学模拟的RMSD轨迹。图中可以看出复合物与蛋白的RMSD轨迹极为接近,复合物的RMSD轨迹大部分被蛋白的RMSD轨迹覆盖。它们的RMSD轨迹测量值范围从0.010至0.050 nm,在25 ns时达到0.040 nm,且在此后的模拟过程中,该波动一直都保持在0.040 nm上下。而配体的RMSD轨迹,测量值范围从0.005至0.030 nm,在5 ns后波动都稳定在0.020 nm左右。这些结果表明该分子动力学轨道具有良好的平衡性,并且系统中的分子对接复合物保持相对稳定。因此,我们采用1号复合物最后10 ns的平均结构来研究,相比于只采用单纯的蛋白质晶体结构而言,会有更好的准确性和稳定性。同时,之前分子对接中得到的复合物也放在一起加以比较(图10b),分子动力学和分子对接中得到的配体分子分别设为橙色和绿色的棍状(图10c)。从图10b中我们可以直观地看到,1号分子在分子动力学模拟与分子对接研究中占据的结合位点相同,且在构型构象上没有明显的差异,这印证了分子对接模型的合理性。但也有一点构象上的改变,仍不能忽视:在结合空腔中,分子动力学中配体的R1和R2取代基与分子对接相比,扭转了一些角度。考虑到可能由于以上的构象变化而带来的作用力的改变,我们对于分子动力学研究中得到复合物的结合机制也进行了相应分析。直观的作用力构成如图11所示,结合作用力主要包括疏水作用和四个氢键作用力。
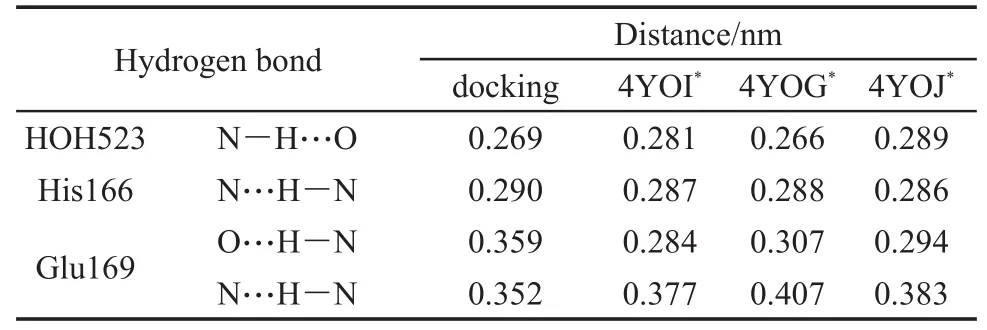
表2 分子对接和实验结果的氢键距离Table 2 H-bond lengths from docking and experiment
从图11中可以得出,1号分子的R2取代基部分固定在一个疏水的口袋之中,这个口袋主要由以下氨基酸残基组成:Gly23、Ser24、Met25、Thr26、Leu49以及Ser147。而R3取代基部分也被一个疏水性的脂肪区域所包裹,组成这个区域的氨基酸主要包括 Phe143、Leu144、Cys145、Gly146、His166、Met168、Glu169以及Leu170。显然,这两个结合区域与分子对接中分析出的疏水位点S1和S2是相一致的。另一方面,分子动力学中配体分子结合的作用力与之前揭示的分子对接中的作用力是较为相似的,结晶水与氨基酸Glu169的重要作用力依然存在,说明了水分子介导的关键作用以及氨基酸Glu169的保守性,在一定程度上支持了分子对接过程中所得到的结论。但由于分子的R1和R2取代基扭转了一些角度的原因使得分子与氨基酸His166的作用力消失,同样与氨基酸Ser24产生了一个新的氢键作用力,使配体更有力地结合于对应位点中,而这并不影响分子整体的结合位点。具体分子对接与分子动力学氢键对比可由表3所示。总之,在水环境下的分子动力学研究中,1号分子在活性位点处保持稳定,所得的结果与分子对接结果也基本保持一致。

图10 对接在3C-like蛋白酶中的1号分子的分子动力学模拟结果Fig.10 MD-simulated results of compound 1 docked in 3C-like protease
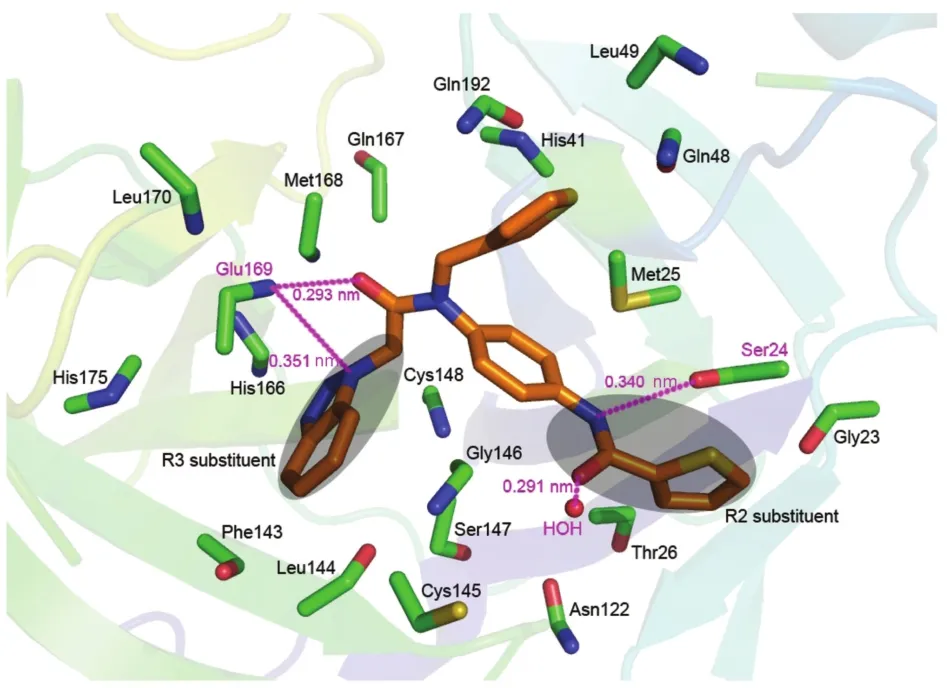
图11 在水环境下分子动力学模拟的结合体的结合位点图Fig.11 Plot of the in-water MD-simulated structures of the binding site

表3 分子对接与分子动力学模拟的氢键分析Table 3 H-bond analysis from docking and MD simulation
3.4.2 2号分子的分子动力学研究
除了1号分子的分子动力学研究,我们还对2号分子进行了50 ns分子动力学研究。2号复合物(黑色)、蛋白(红色)与2号配体(蓝色)的RMSD图以及结合模式图如图12所示。在图12a所示的RMSD图谱中,复合物与蛋白的RMSD数值在10 ns后稳定在0.040 nm左右,而配体的数值则在20 ns后保持在0.020 nm上下,这显示了一个稳定的分子动力学轨道。2号复合物最后10 ns的结合模式图如图12b所示,2号分子的R2取代基部分同样固定在一个由Ser24、Met25、Thr26、Ala46、Asp47以及Leu49等氨基酸残基组成的疏水口袋之中。而R3取代基部分也被一个由 Cys145、Gly146、Cys148、Met168、Glu169、Leu170以及His194等氨基酸组成的疏水脂肪区域所包裹。很显然,这两个小的疏水空腔与前文的位点S1和S2是基本相同的。这表明2号分子仍处于上述提及的疏水结合位点中。
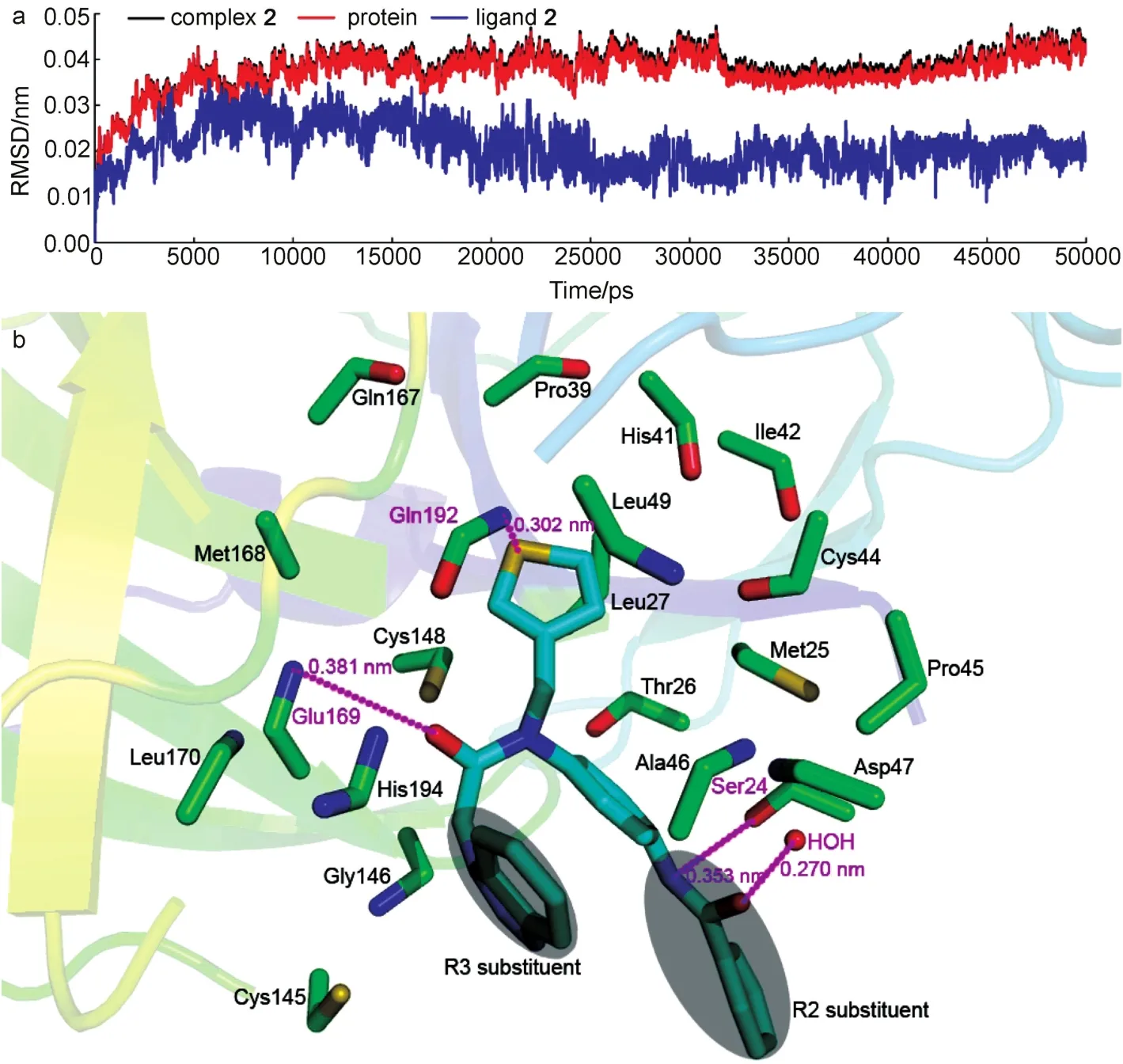
图12 对接在3C-like蛋白酶中的2号分子的分子动力学模拟结果Fig.12 MD-simulated results of compound 2 docked in 3C-like protease
除了疏水作用,2号分子的分子动力学所得到的氢键作用列于表4中,其中也列出了1号分子的分子动力学中的氢键作用。对比表4的数据可以发现,它们各自四个氢键作用中有三个氢键相同,并且可以发现2号分子与Gln192残基又有一个新的氢键作用。分子在结构方面的特征最终决定了它们各自动力学模拟时的特殊构造要求52。我们通过对比1号与2号分子结构之间的差异进行分析,它们的结构特征图如图13所示。由图13可以看出,它们的R1、R3与R4取代基以及R2取代基靠近分子骨架部分拥有共同的结构,而它们的差别仅于R2取代基远位部分,1号分子被噻吩环取代,而2号分子被苯环取代。大部分共同的结构使它们处于位阻、静电、氢键供体与氢键受体场的积极区域有诸多相同,但是由于噻吩环中具有一个S原子可作为氢键受体益于分子活性,导致1号分子活性略高于2号分子。因此,以上1号与2号分子结构之间的微小差异导致了结合模式中氢键的细微改变,体现了R2取代基远位部分在配体与蛋白结合过程中的重要性。综上所述,在1号与2号分子的分子动力学研究中,配体分子在活性位点都保持稳定,所得的两个结果也基本保持一致。
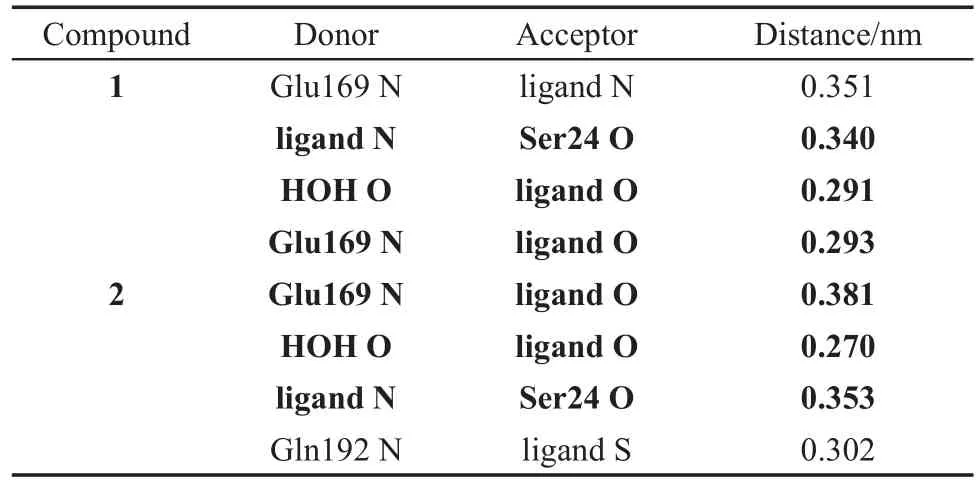
表4 1号与2号分子的分子动力学模拟的氢键分析Table 4 H-bond analysis from MD simulation of compound 1 and compound 2
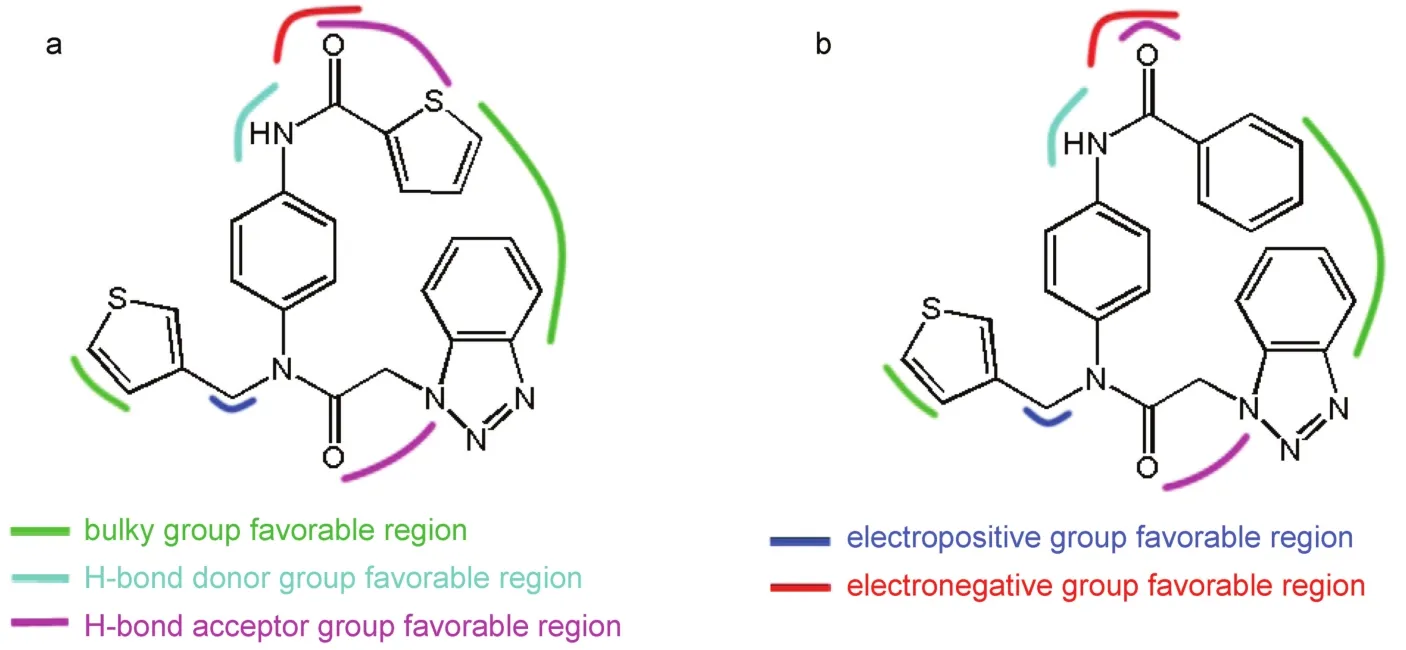
图13 1号与2号分子的结构特征图Fig.13 Structural features of compound 1 and compound 2
3.5新肽类化合物的设计与活性预测
基于43个肽类抑制剂对受体3C-like蛋白酶的抑制活性,建立了具有较强可靠性和良好预测能力的3D-QSAR模型。通过对模型的分析揭示了这类抑制剂分子结构上的特点,为更好地理解配体抑制剂和受体蛋白的作用机理提供了有效的帮助,同时为进一步优化此类抑制剂提供了可靠的依据。图14显示的是我们借助上述研究所获得的基于3C-like蛋白酶上影响肽类分子抑制作用的关键性结构特征。该图将有助于指导筛选和开发更优良的抗MERS-CoV药物。
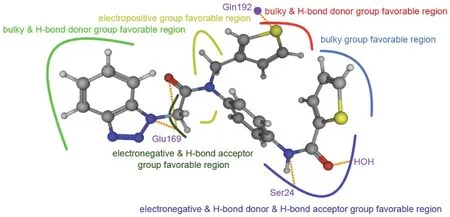
图14 基于3C-like蛋白酶受体上肽类配体分子的交互特性Fig.14 Interaction features of peptidomimetic ligand molecule with the 3C-like protease receptor
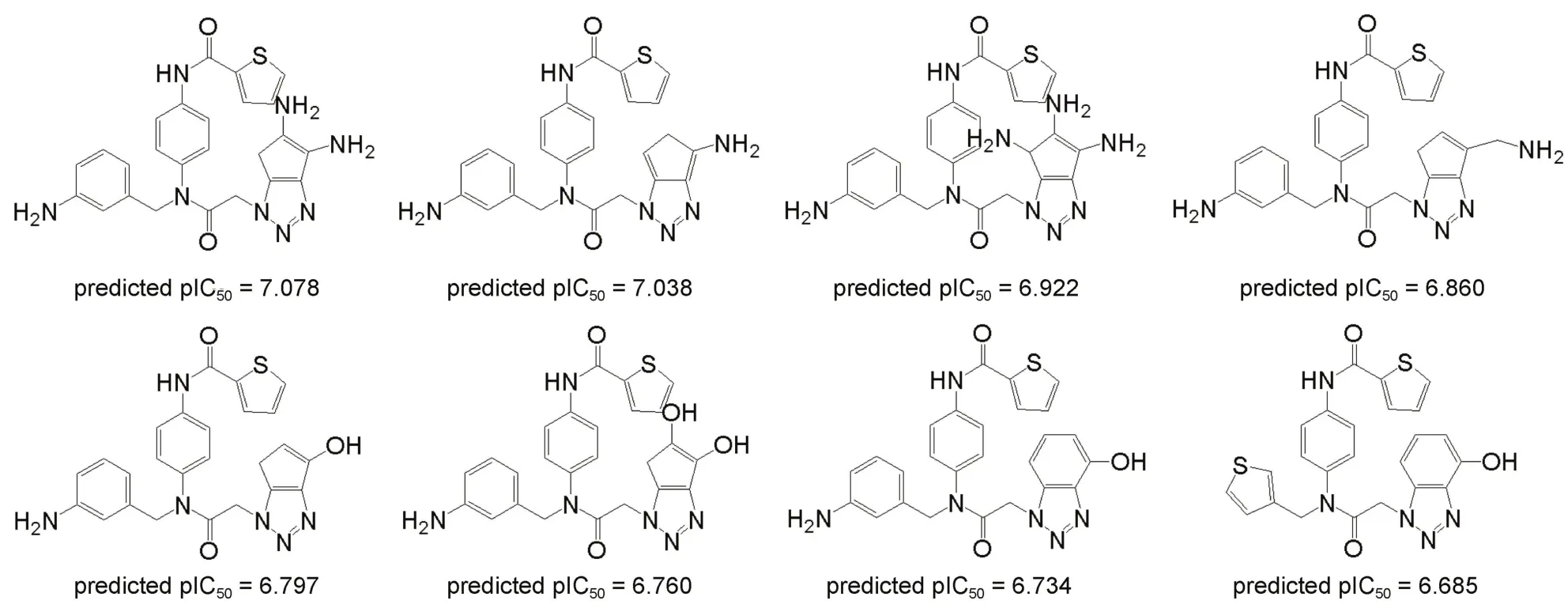
图15 新设计分子的结构Fig.15 Structures of newly designed molecules
例如,以1号分子为模板,我们根据以上肽类分子的交互特性图,通过修饰其各个取代基,设计了8个新的肽类3C-like蛋白酶抑制剂。这8个分子的分子结构如图15所示,CoMSIA模型预测它们的pIC50值均高于模板分子(6.48),关于其具体的药物活性还需通过实验进一步验证。
4 结论
以计算机辅助药物设计方法的理论和手段,借助3D-QSAR建模、分子对接和分子动力学方法对一系列新合成的肽类3C-like蛋白酶抑制剂进行了分子构效关系以及配受体结合作用的研究。本文具体研究结论如下:
(1)建立了基于配体叠合的3D-QSAR模型,模型表现出较好的内部一致性,并得到了CoMSIA模型等统计指标;
(2)对于R1取代基,远离分子骨架的部分(靠近R2取代基的周围),大位阻基团和氢键供体基团有利于分子活性的提高;
(3)对于R2取代基,临近分子骨架的部分,负电性基团、氢键供体基团和氢键受体基团有利于分子活性的提高;同样,靠近R1取代基的周围,大位阻基团也有利于分子活性的提高;
(4)对于R3取代基,临近分子骨架的部分,负电性基团和氢键受体基团有利于分子活性的提高;同样,远离分子骨架的部分,大位阻基团和氢键供体基团也有利于分子活性的提高;
(5)对于R4取代基,临近分子骨架的部分,正电性取代基有利于分子活性的提高;
(6)分子对接结果显示,抑制剂分子通过疏水作用以及与结晶水HOH523、氨基酸His166和Glu169产生的四个氢键作用使小分子稳定在受体的空腔中,说明疏水性以及结晶水、氨基酸His166和Glu169在配体和受体结合过程中产生重要的作用;
(7)分子动力学结果不仅与分子对接结果有较一致的结合空腔,证明了分子对接结果的可靠性,而且在结合空腔中,发现了两个新的关键氨基酸Ser24与Gln192,它们与配体产生了两个较强的氢键。
结论表明我们的3D-QSAR、分子对接和分子动力学的研究得到了一个令人满意的结果。此外,根据这些结论,一些新的具有潜在抑制活性的肽类化合物作为3C-like蛋白酶抑制剂被获得。通过以上结论不但能够深入了解肽类抑制剂的结构特征以及与3C-like蛋白酶的结合作用,也为今后它的实验设计以及抗MERS-CoV药物合成提供了新的方向。
Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Perlman,S.;Netland,J.Nat.Rev.Microbiol.2009,7,439. doi:10.1038/nrmicro2147
(2) Woo,P.C.;Huang,Y.;Lau,S.K.;Yuen,K.Y.Viruses 2010,2, 1804.doi:10.3390/v2081803
(3) Zaki,A.M.;Van Boheemen,S.;Bestebroer,T.M.;Osterhaus, A.D.;Fouchier,R.A.N.Engl.J.Med.2012,367,1814. doi:10.1056/NEJMoa1211721
(4) Rasmussen,S.A.;Gerber,S.I.;Swerdlow,D.L.Clin.Infect. Dis.2015,60,1686.doi:10.1093/cid/civ118
(5) Lau,S.K.;Li,K.S.;Tsang,A.K.;Lam,C.S.;Ahmed,S.; Chen,H.;Chan,K.H.;Woo,P.C.;Yuen,K.Y.J.Virol.2013, 87,8638.doi:10.1128/JVI.01055-13
(6) Chan,J.F.W.;Lau,S.K.P.;Woo,P.C.Y.J.Formos.Med. Assoc.2013,112,372.doi:10.1016/j.jfma.2013.05.010
(7) de Groot,R.J.;Baker,S.C.;Baric,R.S.;Brown,C.S.; Drosten,C.;Enjuanes,L.;Fouchier,R.A.;Galiano,M.; Gorbalenya,A.E.;Memish,Z.A.J.Virol.2013,87,7790. doi:10.1128/JVI.01244-13
(8) Haagmans,B.L.;Al Dhahiry,S.H.;Reusken,C.B.;Raj,V.S.; Galiano,M.;Myers,R.;Godeke,G.J.;Jonges,M.;Farag,E.; Diab,A.Lancet.Infect.Dis.2014,14,140.doi:10.1016/S1473-3099(13)70690-X
(9) Niemeyer,D.;Zillinger,T.;Muth,D.;Zielecki,F.;Horvath,G.; Suliman,T.;Barchet,W.;Weber,F.;Drosten,C.;Müller,M.A. J.Virol.2013,87,12489.doi:10.1128/JVI.01845-13
(10) Matthews,K.L.;Coleman,C.M.;van der Meer,Y.;Snijder,E. J.;Frieman,M.B.J.Gen.Virol.2014,95,874.doi:10.1099/ vir.0.062059-0
(11) Wang,Q.;Qi,J.;Yuan,Y.;Xuan,Y.;Han,P.;Wan,Y.;Ji,W.;Li, Y.;Wu,Y.;Wang,J.Cell Host Microbe 2014,16,328. doi:10.1016/j.chom.2014.08.009
(12) Woo,P.C.;Lau,S.K.;Li,K.S.;Tsang,A.K.;Yuen,K.Y. Emerg.Microbes.Infect.2012,1,e35.doi:10.1038/emi.2012.45
(13) Ziebuhr,J.;Snijder,E.J.;Gorbalenya,A.E.J.Gen.Virol.2000, 81,853.doi:10.1099/0022-1317-81-4-853
(14) Thiel,V.;Ivanov,K.A.;Putics,A.;Hertzig,T.;Schelle,B.; Bayer,S.;Weißbrich,B.;Snijder,E.J.;Rabenau,H.;Doerr,H. W.J.Gen.Virol.2003,84,2305.doi:10.1099/vir.0.19424-0
(15) Liu,W.;Zhu,H.M.;Niu,G.J.;Shi,E.Z.;Chen,J.;Sun,B.; Chen,W.Q.;Zhou,H.G.;Yang,C.Bioorg.Med.Chem.2014, 22,292.doi:10.1016/j.bmc.2013.11.028
(16) Kuo,C.J.;Liang,P.H.ChemBioEng Rev.2015,2,118. doi:10.1002/cben.201400031
(17) Chen,S.;Chen,L.;Tan,J.;Chen,J.;Du,L.;Sun,T.;Shen,J.; Chen,K.;Jiang,H.;Shen,X.J.Biol.Chem.2005,280,164. doi:10.1074/jbc.M408211200
(18) Ramajayam,R.;Tan,K.P.;Liu,H.G.;Liang,P.H.Bioorg. Med.Chem.Lett.2010,20,3569.doi:10.1016/j. bmcl.2010.04.118
(19) Thanigaimalai,P.;Konno,S.;Yamamoto,T.;Koiwai,Y.; Taguchi,A.;Takayama,K.;Yakushiji,F.;Akaji,K.;Chen,S.E.; Naser-Tavakolian,A.Eur.J.Med.Chem.2013,68,372. doi:10.1016/j.ejmech.2013.07.037
(20) Kang,C.M.;Zhao,X.H.;Wang,X.Y.;Cheng,J.G.;Lü,Y.T. Acta Phys.-Chim.Sin.2013,29,431.[康从民,赵绪浩,王新宇,程家高,吕英涛.物理化学学报,2013,29,431.]doi:10.3866/ PKU.WHXB201211151
(21) Zhang,S.Z.;Zheng,C.;Zhu,C.J.Acta Phys.-Chim.Sin.2015, 31,2395.[张淑贞,郑 超,朱长进.物理化学学报,2015,31, 2395.]doi:10.3866/PKU.WHXB201510142
(22) Pillaiyar,T.;Manickam,M.;Jung,S.H.Med.Chem.2015,5, 361.doi:10.4172/2161-0444.1000287
(23) Ren,Z.;Yan,L.;Zhang,N.;Guo,Y.;Yang,C.;Lou,Z.;Rao,Z. Protein Cell 2013,4,248.doi:10.1007/s13238-013-2841-3
(24) Deng,X.;StJohn,S.E.;Osswald,H.L.;O'Brien,A.;Banach,B. S.;Sleeman,K.;Ghosh,A.K.;Mesecar,A.D.;Baker,S.C.J. Virol.2014,88,11886.doi:10.1128/JVI.01528-14
(25) Tomar,S.;Johnston,M.L.;John,S.E.S.;Osswald,H.L.; Nyalapatla,P.R.;Paul,L.N.;Ghosh,A.K.;Denison,M.R.; Mesecar,A.D.J.Biol.Chem.2015,290,19403.doi:10.1074/ jbc.M115.651463
(26) John,S.E.S.;Tomar,S.;Stauffer,S.R.;Mesecar,A.D.Bioorg. Med.Chem.2015,23,6036.doi:10.1016/j.bmc.2015.06.039
(27) AbdulHameed,M.D.M.;Hamza,A.;Liu,J.J.;Zhan,C.G.J. Chem.Inf.Model.2008,48,1760.doi:10.1021/ci800147v
(28) Gasteiger,J.;Marsili,M.Tetrahedron 1980,36,3219. doi:10.1016/0040-4020(80)80168-2
(29) Clark,M.;Cramer,R.D.;Van Opdenbosch,N.J.Comput. Chem.1989,10,982.doi:10.1002/jcc.540100804
(30) Cramer,R.D.;Patterson,D.E.;Bunce,J.D.J.Am.Chem.Soc. 1988,110,5959.doi:10.1021/ja00226a005
(31) Klebe,G.;Abraham,U.;Mietzner,T.J.Med.Chem.1994,37, 4130.doi:10.1021/jm00050a010
(32) Edraki,N.;Das,U.;Hemateenejad,B.;Dimmock,J.R.Iran.J. Pharm.Res.2016,15,425.
(33) Li,X.L.;Ye,L.;Wang,X.X.;Wang,X.Z.;Liu,H.L.;Qian,X. P.;Zhu,Y.L.;Yu,H.X.Sci.Total Environ.2012,441,230. doi:10.1016/j.scitotenv.2012.08.072
(34) Shah,P.;Saquib,M.;Sharma,S.;Husain,I.;Sharma,S.K.; Singh,V.;Srivastava,R.;Shaw,A.K.;Siddiqi,M.I.Bioorg. Chem.2015,59,91.doi:10.1016/j.bioorg.2015.02.001
(35) Zhang,S.;Hou,B.;Yang,H.;Zuo,Z.Arch.Pharm.Res.2016, 1.doi:10.1007/s12272-016-0709-9
(36) Berman,H.M.;Battistuz,T.;Bhat,T.N.;Bluhm,W.F.;Bourne, P.E.;Burkhardt,K.;Iype,L.;Jain,S.;Fagan,P.;Marvin,J.; Padilla,D.;Ravichandran,V.;Schneider,B.;Thanki,N.; Weissig,H.;Westbrook,J.D.;Zardecki,C.Acta Crystallogr.D 2002,58,899.doi:10.1107/S0907444902003451
(37) Verdonk,M.L.;Cole,J.C.;Hartshorn,M.J.;Murray,C.W.; Taylor,R.D.Proteins 2003,52,609.doi:10.1002/prot.10465
(38) Duan,A.X.;Chen,J.;Liu,H.D.;Liu,X.H.;Lu,X.Q.J.Mol. Sci.2009,25,473.[段爱霞,陈 晶,刘宏德,刘秀辉,卢小泉.分析科学学报,2009,25,473.]
(39) Arooj,M.;Sakkiah,S.;Kim,S.;Arulalapperumal,V.;Lee,K. W.PloS One 2013,8,e63030.doi:10.1371/journal. pone.0063030
(40) Van der Spoel,D.;Lindahl,E.;Hess,B.;Groenhof,G.;Mark,A. E.;Berendsen,H.J.C.J.Comput.Chem.2005,26,1701. doi:10.1002/jcc.20291
(41) Lindahl,E.;Hess,B.;Van Der Spoel,D.J.Mol.Model.2001,7, 306.doi:10.1007/s008940100045
(42) vanAalten,D.M.F.;Bywater,R.;Findlay,J.B.C.;Hendlich, M.;Hooft,R.W.W.;Vriend,G.J.Comput.Aid.Mol.Des. 1996,10,255.doi:10.1007/BF00355047
(43) Berendsen,H.J.C.;Postma,J.P.M.;van Gunsteren,W.F.; Hermans,J.Interaction Models for Water in Relation to Protein Hydration.In Intermolecular Forces;Springer Netherlands: Berlin,1981;pp 331-342.
(44) Ryckaert,J.P.;Ciccotti,G.;Berendsen,H.J.C.J.Comput. Phys.1977,23,327.doi:10.1016/0021-9991(77)90098-5
(45) Essmann,U.;Perera,L.;Berkowitz,M.L.;Darden,T.;Lee,H.; Pedersen,L.G.J.Chem.Phys.1995,103,8577.doi:10.1063/ 1.470117
(46) Alexander,G.;Alexander,T.J.Mol.Graph.Model.2002,20, 269.doi:10.1016/S1093-3263(01)00123-1
(47) Kamsri,P.;Punkvang,A.;Hannongbua,S.;Saparpakorn,P.; Pungpo,P.RSC Adv.2015,5,52926.doi:10.1039/C5RA08103C
(48) Gao,X.;Han,L.;Ren,Y.Molecules 2016,21,591.doi:10.3390/ molecules21050591
(49) Da,C.X.;Mooberry,S.L.;Gupton,J.T.;Kellogg,G.E. J.Med.Chem.2013,56,7382.doi:10.1021/jm400954h
(50) Jeffrey,G.A.An Introduction to Hydrogen Bonding.In Topics in Physical Chemistry;Oxford University Press:NewYork,1997.
(51) Minini,L.;Alvarez,G.;González,M.;Cerecetto,H.;Merlino, A.J.Mol.Graph.Model.2015,58,40.doi:10.1016/j. jmgm.2015.02.002
(52) Wang,J.;Li,F.;Li,Y.;Yang,Y.;Zhang,S.;Yang,L.Mol. BioSyst.2013,9,2296.doi:10.1039/c3mb70105k
QSAR,Molecular Docking and Molecular Dynamics of 3C-like Protease Inhibitors
LIN Feng1FU Xin-Mei2,*WANG Chao1JIANG Si-Yu1WANG Jing-Hui1ZHANG Shu-Wei1YANG Ling3LIYan1,*
(1School of Chemical Engineering,Dalian University of Technology,Dalian 116024,Liaoning Province,P.R.China;2State Key Laboratory of Fine Chemicals,Dalian University of Technology,Dalian 116024,Liaoning Province,P.R.China;3Laboratory of Pharmaceutical Resource Discovery,Dalian Institute of Chemical Physics,Chinese Academy of Sciences, Dalian 116023,Liaoning Province,P.R.China)
3C-like protease is an extremely important protease involved in the multiplicative process of coronaviruses,includingthedeadlyMiddleEast respiratorysyndromecoronavirus(MERS-CoV).3C-likeprotease has become a hot research topic in the field of coronavirology.For the first time,a set of ligand-and receptorbased three-dimensional quantitative structure-activity relationships(3D-QSAR)models were carried out via comparative molecular field analysis(CoMFA)and comparative molecular similarity indices analysis(CoMSIA)to explore the structure-activity correlation of 43 peptidomimetic inhibitors of the 3C-like protease of the bat coronavirus HKU4(HKU4-CoV),which belongs to the same 2c lineage as MERS-CoV and shows high sequence similarity with MERS-CoV.Based on the ligand-based alignment,an optimal CoMSIAmodel(yielded by steric, electrostatic,H-bond donor and H-bond acceptor fields)was obtained with good predictive power of Q2=0.522, R2ncv=0.996 and R2pre=0.904(Q2:cross-validated correlation coefficient,R2ncv:non-cross-validated correlation coefficient,R2pre:predicted correlation coefficient for the test set of compounds).Molecular docking and molecular dynamics simulations were performed according to this model to further determine the interaction mechanism between ligands and the receptor.The experimental results show:(1)based on the optimal CoMSIAmodel,the 3D contour maps vividly illustrate that the molecular biological activity is influenced by the steric,electrostatic, H-bond donor and H-bond acceptor interactions of molecular groups.(2)Based on the docking analysis, hydrophobicity,crystal water,His166andGlu169haveimportantrolesintheligandsandreceptorbindingprocess. (3)Molecular dynamics(MD)simulations were carried out for further verification of the reliability of the docking model,and provide two new key residues,Ser24 and Gln192,which have two strong hydrogen bonds with the ligands.Some new compounds were obtained based on the modeling that are potential peptidomimetic inhibitors of 3C-like protease.These results help establish the binding mechanism between 3C-like protease and peptidomimetic inhibitors,and provide a valuable reference for future anti-MERS-CoV drug design.
MERS-CoV;3C-like protease;Peptidomimetic inhibitor;3D-QSAR;Molecular docking; Molecular dynamics
O641
10.3866/PKU.WHXB201608121
Received:May 31,2016;Revised:August 9,2016;Published online:August 12,2016.
*Corresponding authors.FU Xin-Mei,Email:fuxinmei@dlut.edu.cn;Tel:+86-411-84986206.LI Yan,Email:yanli@dlut.edu.cn; Tel:+86-411-84986062.
The project was supported by the National Natural Science Foundation of China(11201049).
国家自然科学基金(11201049)资助项目
