美国、欧盟、日本细胞治疗监管政策研究
2016-12-22吴曙霞杨淑娇吴祖泽
吴曙霞,杨淑娇,吴祖泽
美国、欧盟、日本细胞治疗监管政策研究
吴曙霞,杨淑娇,吴祖泽
生物治疗作为外科手术、药物治疗、放射治疗之后的第四大治疗技术,已经得到全球的广泛认同。生物治疗主要由细胞治疗、基因治疗、组织工程等领域组成,其中细胞治疗由于发展的快速性和重要性,引起了科学界与临床应用的更多关注。作为一个冉冉升起的新兴医药产品领域,细胞治疗受到了全球发达国家政府的战略重视和投入,也成为产品研发和风险投资关注的热点领域。目前以干细胞治疗和免疫细胞治疗为主流的细胞治疗产品在人类疾病治疗中的地位和价值已经初步显现,尤其是在众多未满足需求的难治性疾病领域,如恶性肿瘤、疑难重症的治疗和严重创伤修复的再生医学领域,细胞治疗已经成为弥补传统治疗不可或缺的有效手段。对于一些传统药物或治疗手段束手无策的重大疾病,如重症肝病、移植物抗宿主病(GvHD)、肾移植排斥、系统性红斑狼疮、帕金森病、老年痴呆等,细胞治疗均显示出了明确的疗效。
在干细胞、基因组编辑、Cart-T 免疫治疗等前沿领域的推动下,细胞治疗产品研究发展非常迅猛,全球细胞治疗领域也面临着科技发展、临床需求与监管审批的矛盾。细胞治疗既是临床疾病治疗的一种个体化先进治疗技术,也可以作为药物大规模生产,各国对于细胞治疗的政策监管各有不同。本文从目前已有产品审批现状入手,从全球的视野分析细胞治疗产品的产业进展,并对产品研发较为领先的美国、欧盟、日本等国的法规政策框架与监管进行梳理,以期为我国细胞治疗类产品的发展提供一些启示与借鉴。
1 全球已经获批的产品现状
依据细胞来源的增殖性,细胞治疗产品可以分为成体细胞与干细胞类产品。成体细胞类产品多为自体细胞,干细胞类产品多为间充质来源,美国、欧盟、日本、韩国、加拿大等国已有产品上市。
1.1 成体细胞产品
上市的成体细胞治疗产品主要集中于美国、欧盟、日本等国(表 1),细胞来源包括自体软骨细胞、表皮细胞、单核细胞、成纤维细胞等,主要适应证集中于骨缺损修复替代、皮肤烧伤、癌症的免疫治疗等。
1.2 干细胞产品
美国、欧盟、澳大利亚、韩国、日本、加拿大等国已经有干细胞上市(表 2),细胞来源包括不同组织来源的自体间充质干细胞、造血干细胞等,异体来源细胞较少。
2 细胞治疗概念辨析与风险
由于细胞治疗技术发展的快速性与尚未被科学界明确的未知风险,如何对细胞治疗领域进行适度的监管和规范也受到各国的重视。以下对已上市产品较多的美国、欧盟与日本等国,从细胞治疗类产品的概念界定和监管风险予以分析。
2.1 产品概念界定
美国将细胞、组织或基于细胞、组织的产品(HCT/Ps)归类监管[1],欧盟以先进技术治疗医学产品(advanced therapy medicinal product,ATMP)归类监管[2],日本则按照再生医学产品管理[3]。美国、欧盟、日本对于细胞治疗产品的概念界定的侧重点各有不同(表 3),美国注重界定给药方式;欧盟侧重于临床应用范围,可用于疾病的预防、诊断或治疗,并且强调了被处理和生物学特性;日本则强调了细胞的来源为自体或同源,对细胞进行的人工基因操作技术和主要用于治疗与再生修复。因此可以归纳出,细胞治疗的产品界定可从细胞的来源、细胞人工操作的技术范围、给药的方式、临床适用的范围进行约束。
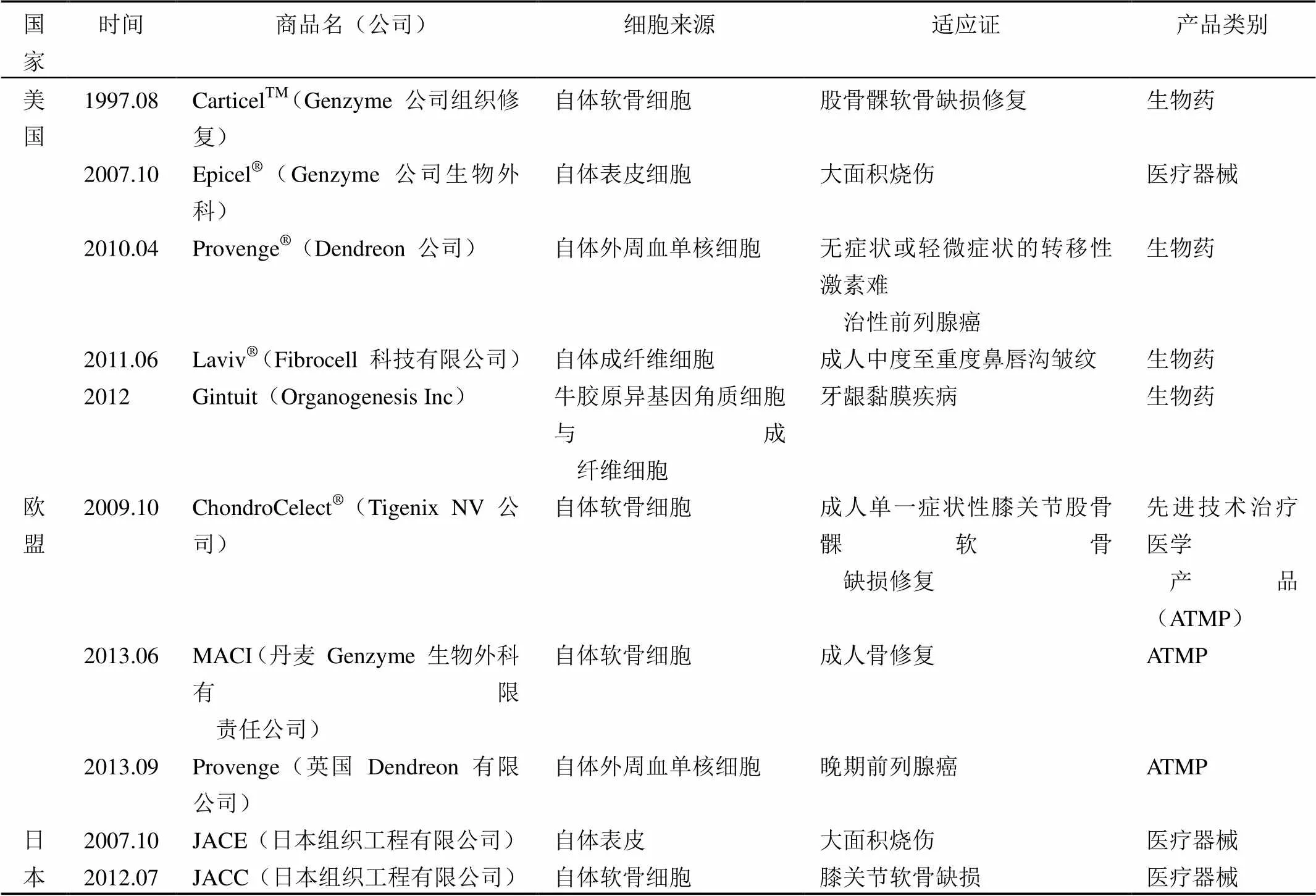
表 1 全球已经上市的成体细胞产品
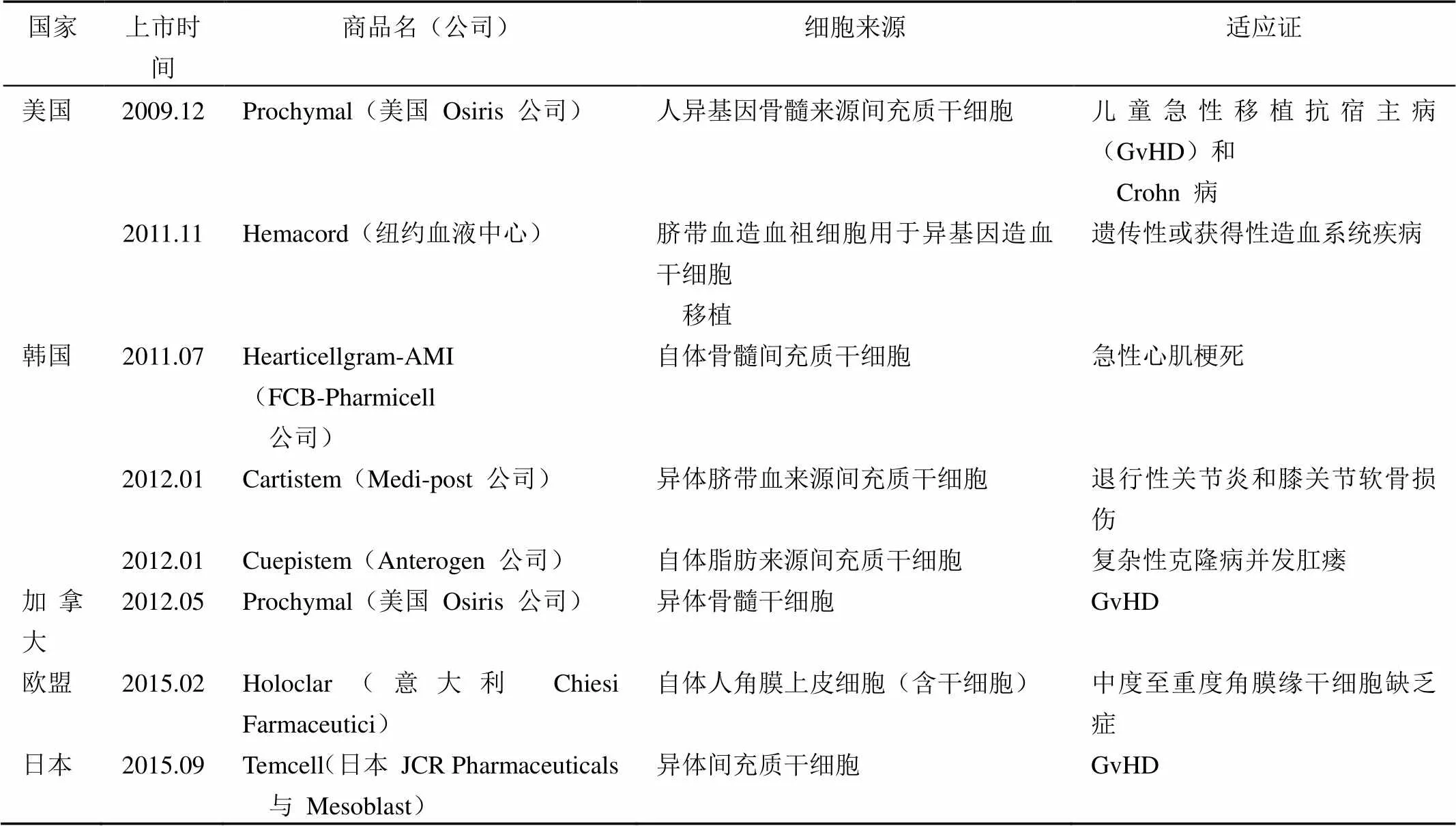
表 2 全球已经上市的干细胞产品

表 3 美国、欧盟、日本对于细胞治疗产品的概念界定
2.2 细胞治疗产品风险考量
细胞治疗产品的类别不同,其临床应用的风险性也有显著差异,同时新型基因工程技术的介入使得细胞治疗领域更为复杂,这使得细胞治疗的监管充满挑战,因此目前各国对于细胞治疗产品上市都呈审慎态度。依据细胞的增殖性,细胞治疗产品可分为干细胞产品与成体细胞产品;成体细胞只具备局部修复能力,但没有增殖的潜力,因此风险性相对较低。干细胞具备增殖的潜力,使得干细胞临床治疗仍存在许多无法避免的危险因素,如干细胞及其衍生物的安全性、纯度、稳定性和有效性难以保证,其次干细胞的自我更新和非目的性分化无法控制,极易形成畸胎瘤或肿瘤;而且干细胞及其衍生物进入人体后如何控制使其不产生异位组织和部位迁移同样非常重要。干细胞中,自体造血干细胞、间充质干细胞、神经干细胞、脂肪干细胞等成体干细胞移植,相对来说比较安全[4]。
据细胞的来源,细胞治疗产品可以分为自体细胞、同源异体细胞、异种细胞等,由于生物体具有排异性,异体与异种细胞在临床治疗中风险相对较高,治疗效果不确定性较高。从已上市细胞治疗产品来看,目前临床上市的大多为自体细胞类产品。
3 细胞治疗审批监管法规政策框架
虽然不同国家卫生与药监管理体系职能有所不同,从监管与审批来看细胞治疗产品与临床应用总体分为两条路径,一是以药物或医疗器械产品由药品监管部门进行临床准入与应用的监管审批,需要严格遵循药物产品审批的流程。二是医疗技术由卫生部门进行监管,在医院直接进行临床应用,不同国家法规界定的细胞治疗类型与应用有所差异。
3.1 美国
美国在细胞治疗领域已经形成了完善的法规监管框架,由上位法律、法规、管理制度与指南三层组成其法律法规体系(表 4)。美国细胞、组织或基于细胞、组织的产品(human cells, tissues, or cellular or tissue-based products, HCT/Ps)属于人类组织和细胞类产品范畴[5],分为 PHS 351 产品与 PHS 361 产品两大类管理,PHS 351 产品由 FDA 的生物制品评估研究中心(CBER)统一负责审批,PHS 361 产品可以在医院直接进行临床应用。
3.1.1 法律法规 从法律层面,细胞治疗管理的法律依据来自于两个国会法案,即《美国食品、药品和化妆品法案》(FD&C Act)及《公共卫生服务法案》(PHS Act)。《美国联邦条例》(CFR)是 FDA 管理要求的构建基础,最适用于干细胞的法规是对提交研究性新药申请要求做出的规定(21CFR 312),以及关于现行药品生产质量管理规范的法规(21CFR 210&211)。《现行药品生产质量管理规范》(cGMP)是根据 CFR 中的 21CFR 210 和 211 条制定的,其中的原则适用于干细胞产品,包括制造产品设备的物理特征及在某种设备中制造细胞产品的过程和步骤。
美国于 2001 年发布 CFR1271 管理法规,并于 2005 年正式实施。这是细胞治疗审批主要依据的法规,将人体细胞组织分为 PHS 351 产品与 PHS 361 产品两大类管理,PHS 351 产品属于 HCT/Ps 分类监管的产品,包括骨、韧带、皮肤、硬脑膜、心脏瓣膜、角膜、外周血干细胞(PBSCs)、脐带血来源前体细胞、经过改造的自体软骨细胞、人工合成基质上的表皮细胞、精子或其他生殖组织。
3.1.2 指南与规范 FDA 还与其他细胞治疗领域管理部门、企业、研究机构相互沟通、相互影响,由此形成了一些有关大多数种类生物产品的制造和临床试验的指南规范[6]。在这些共同制定的大量指导性文件中,界定的原则适用于细胞疗法的评估。FDA 和 NIH 之间通过签署正式的谅解备忘录(MOU)协议促进干细胞管理建议的形成。
3.1.3 审批监管框架 FDA 生物制品评估研究中心下设细胞、组织与基因治疗办公室,该办公室由人类组织管理、临床评估与药理、细胞与基因治疗三个部门组成,其中细胞与基因治疗部负责接收细胞治疗产品的审批与准入,快速审批程序时间为6 ~ 10 个月,目前美国已有多种细胞治疗产品上市。

表 4 美国细胞治疗产品主要监管法律法规与指南规范
PHS 361 类细胞治疗产品无需向 FDA 提出IND 申请,不属于 HCT/Ps 分类监管的产品,包括全血、血液成分或血液衍生产品,如白细胞、血小板、凝血因子等(由血液制品相关规定监管)动物来源的细胞、组织或器官干预最小化、作为同源性应用的骨髓等。PHS 361 产品需要同时满足以下条件:制备过程符合干预最小化,仅同源性使用,未添加水、晶体液或杀菌、保存、存储剂之外的其他任何试剂。并且不产生全身反应,且不依赖活体细胞的代谢过程发挥作用;若产生全身反应或依赖活体细胞的代谢过程发挥作用,则必须是符合作用同源性且用于 1、2 级亲属的异基因移植或作为生殖应用。其中产品最小化干预(minimally manipulated)是指在细胞的处理过程中,不能改变相关的生理特性(即未经过体外激活、包裹、扩增或基因修饰等)。
3.2 欧盟
欧盟药品管理局将组织工程、细胞治疗、基因治疗产品纳入先进技术治疗医学产品(ATMP)管理,该类药物的定义是能够为疾病带来革命性的治疗方案,对于患者与产业具有巨大前景。欧盟细胞治疗的监管有两条路径。一是按照先进技术治疗医学产品进行临床研究与申报,由欧洲药品管理局负责审批和管理。二是遵循医院豁免条款,由医院决定对患者的治疗应用。
3.2.1 法律法规 从法律层面,细胞治疗管理的法律依据为欧盟《医药产品法》与《医疗器械法》,对医药产品的临床前研究、临床研究、制造与销售进行全产业链的系统提供法律监管框架[7](表 5)。随着生物治疗的飞速发展,欧盟加强了对细胞治疗产品的监管,2007 年欧盟颁布了《先进技术治疗医学产品法规》(Regulation (EC) No. 1397/2007 on Advanced Therapy Medicinal Product),于 2008 年12 月 30 日起实施,将基因治疗产品、体细胞治疗产品和组织工程产品定义为先进技术治疗医学产品,其中细胞治疗产品指含有经过处理的被改变了生物学特性的细胞或者组织,可以用于疾病的治疗、诊断或者预防。按照药物申报,先进技术治疗医学委员会审批,审批时间 1 ~ 2 年。该法规中提出了医院豁免条款(Article 28),对某一医生进行的,为患者个体进行的治疗应用行为进行豁免。该条款允许欧洲医院在经过基础研究、临床研究验证有效性与安全性之后,可以生产小规模的细胞产品用于特定的患者,主要是临床中心进行自体细胞治疗。

表 5 欧盟细胞治疗产品主要监管法律法规与指南规范
3.2.2 指南与规范 欧盟针对基因治疗和细胞治疗产品还制定了一系列科学指导原则。这些指导原则提出了对 ATMP 的研发和监管要求,如基于风险的产品开发途径和评价理念、对于细胞和结构组分之间相互作用的特殊要求、对于临床/非临床的灵活性考虑、对于药品临床试验管理规范(GCP)的特殊要求,以及关于上市后安全有效性跟踪和风险管理的特殊考虑等。
3.2.3 审批监管框架 在审评程序方面,欧盟规定 ATMP 必须执行集中化审评程序,并成立了先进技术疗法委员会(CAT),专门负责新技术疗法产品的技术审评。CAT 对每一份提交至管理部门的ATMP 提出审评意见,但该意见将被提交至人用医药产品委员会(CHMP),由 CHMP 做出采纳批准、变更、暂停或取消上市许可的建议,然后将建议发送至欧盟委员会做决定。一旦产品在欧盟被批准上市,管理部门将对其安全性和有效性进行进一步评价。同时,为鼓励 ATMP 的研究和开发,欧盟执行了一些特殊的支持鼓励政策,如减免申请人向管理部门支付的部分费用、申请人可从欧盟获得更多科学支持和帮助等。
3.3 日本
日本政府正在实行举国战略,相继修订并出台了一系列有关再生医学的新法规(表 6),建立更加高效的通道将促进细胞技术转化到临床应用,以保证日本在再生医学领域的研究治疗优势。日本将细胞治疗、基因治疗、组织工程作为独立于药物、医疗器械的再生医学产品单独监管[8],并在 2013 年进行了再生医学产品的审批改革。

表 6 日本细胞治疗产品主要监管法律法规与指南规范
3.3.1 法律法规 日本 2013 年修订了药事法,将其更名为药物、医疗器械与其他产品法,于 2014 年 11 月实施[9],修订增加了再生医学产品监管的部分。日本国会也认识到现有监管体制在细胞治疗领域的缺失,在 2013 – 2014 年相继出台了再生医学促进法与再生医学安全法,从研发与临床应用方面提供了法规依据。
3.3.2 指南规范 日本出台了一系列研究指南规范,包括《干细胞临床研究指南》、《人体自体细胞/组织产品质量控制与安全指南》、《细胞组织操作原则》等。日本政府也在考虑对细胞治疗的监管立法建立分级管理制度,针对诱导多功能干细胞、间充质干细胞、免疫细胞治疗分别制定不同级别的管理办法。
3.3.3 审批监管框架 日本再生医学产品由日本药品医疗器械管理局依据《药物、医疗器械与其他产品法》进行监管,其药品评估中心下设细胞与组织类产品审批办公室负责具体审批事务。再生医学产品在原有药物审批程序基础研究、临床研究、临床试验、审批准入的基础上,在临床研究证实再生医学产品的有效性与安全性之后,增加了条件性限制性准入许可。再生医学产品需要满足以下条件:适应证为危及生命的疾病,治疗方法为满足需求的创新性产品,并经过初步的有效性、安全性验证,符合相关监管法规政策。在经过患者知情同意后产品进入市场,大大加快了产品临床应用的进程。
条件性限制性准入许可时间最长为 7 年,在证明细胞治疗产品临床试验与应用有效性之后,产品可以申请作为正式的再生医学产品长期上市,7 年时间到期后再次进行申请或者退出市场。目前已有一种用于缺血性心脏病严重心衰的骨骼肌细胞产品通过条件性限制性准入许可进入市场,市场考察期为 5 年。
在医院实施的细胞治疗,日本负责医疗卫生和社会保障的主要部门厚生劳动省,依照 2014 年新实施的《再生医学安全法》对细胞治疗进行监管。监管范围包括所有未经证实其安全有效性的使用细胞治疗的医疗技术,目前日本已经批准了 40 家具有细胞治疗资质的研究中心,主要面向由研究者进行的临床研究与类似欧盟的医院豁免类细胞治疗应用。
4 结论与讨论
目前以细胞治疗为代表的生物治疗虽然不是临床疾病救助的主流治疗措施,但从各国的发展来看,生物治疗、细胞治疗已经成为重要的药物与医疗技术的组成部分,相关科学问题正逐渐阐明,越来越多的治疗产品进入产业化,细胞治疗产业也正由无序走向有序。细胞治疗已成为各国政府、科技和企业界高度关注和大力投入的重要研究发展领域,也是代表国家科技实力的战略必争领域。
4.1 细胞治疗领域需要建立系统的监管与政策框架
从美国、欧洲、日本等国的经验,细胞治疗领域有必要建立从法律、法规到行业指南不同层次的监管与政策框架,分级分类管理,促进细胞治疗的医疗技术临床转化。虽然临床治疗的场所主要发生在医院,但临床治疗产品的监管和准入,从国际惯例而言主要由药品监督部门承担,而卫生部门主要承担对医师、临床研究医疗技术应用的监管。欧盟、日本等国经验表明政府积极的态度应对技术发展给监管带来的挑战,积极进行修订监管法规[10],并出台了针对再生医学与细胞治疗产品的相关法案,颁布大量指南与规范指导产品研发和质量控制,有助于保障与提升相关领域技术与产业在全球的领先性。
4.2 细胞治疗的发展需要产学研协同应对监管挑战
细胞治疗产业的快速发展势必改变全球生物医药产业格局,是我国医药产业升级发展的重大战略机遇。对于奠定我国在未来全球医学领域的战略地位,引领医药科技发展、保障我国国民健康至关重要。我国近 5 年在干细胞及相关领域科研经费投入总额超过 10 亿元,对干细胞的基础研究、关键技术和资源平台建设给予了大力支持,取得了一批标志性成果。但在细胞治疗领域,我国目前存在很大的转化瓶颈,至今还没有产品上市,未来我国生物治疗领域的发展不仅需要科研机构、医药产业、医疗机构与政府监管的合力,而且还需要专业生物医药智库的支撑,以提出我国不同阶段的政策框架与发展路线图,推动我国细胞治疗领域健康有序的发展。
4.3 我国亟需加强法规、政策与执行体系的系统建设
从法规治理层面,我国药监与卫生部门已着手出台相关政策法规,但目前在细胞治疗领域遵循的法规政策与审批路径欠清晰,并且难以覆盖从科研、产品研发、临床应用的全流程。虽然我国细胞治疗领域的基础研究能力已经达到与发达国家“并跑”的水平,但监管与审批准入已经成为产业发展的瓶颈,制约了产业化与临床应用的发展。建议我国完善相关法规体系,明确分工与管理路径。设立专门负责细胞药品的受理、咨询和审评部门,建立细胞药品的申报审批路径,完善审评标准,加快创新药品的审批。制定细胞医疗技术分类分级审批流程和监管措施等,明确临床转化的路径和标准。如成熟的自体免疫细胞治疗技术和已经经过临床研究证明安全性好、疗效确切且传统治疗不可替代的自体干细胞医疗技术,经立项与评审后,可择优在经批准的临床研究基地或医院应用于临床救治。
[1] U.S. Food & Drug Administration. Human cells, tissues, and cellular and tissue-based products, 21CFR1271. 2012 [2016-08-10]. http:// www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart¼1271.
[2] The European Parliament and of the Council. On advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. 2007(2007-11-13) [2016-09-12]. http://ec.europa.eu/health/files/eudralex/vol-1/reg_2007_1394_cons_2012-07/reg_2007_1394_cons_2012-07_en.pdf.
[3] Konishi A, Sakushima K, Isobe S, et al. First approval of regenerative medical products under the PMD act in Japan. Cell Stem Cell, 2016, 18(4):434-435.
[4] Hu ZB, Wang LS, Cui CP, et al. The clinical safety assessment report of stem cell therapy. Chin Med Biotechnol, 2013, 8(5):349-361. (in Chinese)
胡泽斌, 王立生, 崔春萍, 等. 干细胞临床应用安全性评估报告. 中国医药生物技术, 2013, 8(5):349-361.
[5] Bailey AM, Arcidiacono J, Benton KA, et al. United States Food and Drug Administration regulation of gene and cell therapies. Adv Exp Med Biol, 2015, 871:1-29.
[6] U.S. Food & Drug Administration. Cellular & gene therapy guidances. [2016-09-15]. http://www.fda.gov/BiologicsBloodVaccines/Guidance ComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/default.htm.
[7] Salmikangas P, Schuessler-Lenz M, Ruiz S, et al. Marketing regulatory oversight of advanced therapy medicinal products (ATMPs) in Europe: the EMA/CAT perspective. Adv Exp Med Biol, 2015, 871:103-130.
[8] Maeda D, Yamaguchi T, Ishizuka T, et al. Regulatory prameworks for gene and cell therapies in Japan. Adv Exp Med Biol, 2015, 871: 147-162.
[9] Okada K, Kazuhisa Koike K, Sawa Y. Consideration of and expectations for the pharmaceuticals, medical devices and other therapeutic products act in Japan. Regenerative Therapy, 2015, 1(6):80-83.
[10] Maciulaitis R, D'Apote L, Buchanan A, et al. Clinical development of advanced therapy medicinal products in Europe: evidence that regulators must be proactive. Mol Ther, 2012, 20(3):479-482.
国家自然科学专项基金(L1222031);北京市科技计划课题(Z151100003115064)
100850 北京,军事医学科学院卫生勤务与医学情报研究所(吴曙霞、杨淑娇);100850 北京,军事医学科学院辐射与放射医学研究所(吴祖泽)
吴祖泽,Email:celiawoo@126.com
2016-10-10
10.3969/j.issn.1673-713X.2016.06.002
