悬浮泥沙在长江口铀非保守行为中的重要作用
2016-12-21周婧杜金洲毕倩倩王锦龙刘丹彤
周婧,杜金洲*,毕倩倩,王锦龙,刘丹彤
(1.华东师范大学 河口海岸学国家重点实验室,上海 200062)
悬浮泥沙在长江口铀非保守行为中的重要作用
周婧1,杜金洲1*,毕倩倩1,王锦龙1,刘丹彤1
(1.华东师范大学 河口海岸学国家重点实验室,上海 200062)
利用高精度的电感耦合等离子体质谱仪对2014年1月长江口表层水中溶解铀浓度及其234U/238U比值、2013年3月长江口表层沉积物中各矿物组分的铀含量及其234U/238U比值进行了测定,研究了其空间分布特征和影响因素。结果表明:除了长江径流和海水之外,长江口还有其他的溶解铀来源。水体中过剩铀与悬浮颗粒物浓度呈现显著相关性(r2=0.96)。对长江口表层沉积物进行的序列提取实验进一步表明,水体中悬浮颗粒物或沉积物中可解吸态和碳酸钙结合态铀可以在河口区域释放进入水体,而铁锰氧化物和有机物结合铀比较稳定,不受河口区混合过程的影响。每千克颗粒物或沉积物能够释放约2 μmol颗粒态铀,使其转化为溶解态。然而,铁氢氧化物和细颗粒物的絮凝吸附作用也可使溶解铀同时从河口水体中清除。在低盐度区,铀的清除和添加过程速率相近,使溶解铀呈现暂时的“伪保守”现象:颗粒态释放的铀具有明显低的234U/238U比值,导致水体的234U/238U低于保守混合值。在中高盐度区域,溶解铀呈现明显的富集现象。但是由于水相和颗粒相中的铀交换,可释放颗粒态铀的234U/238U接近溶解铀的234U/238U比值,从而导致水体的234U/238U比值呈现出保守性。长江口颗粒物的铀释放通量为(3.48±0.41)×105mol/a,约占输入的总颗粒态铀通量(1.80±0.17)×106mol/a的19.3%。长江口输入东海的溶解铀总通量(河流溶解态铀与河口添加铀之和)为(2.68±0.13)×106mol/a,约为世界河流入海铀通量的11.7%。
溶解铀;颗粒态铀;234U/238U 比值;非保守;长江口
1 引言
利用铀系核素放射性不平衡特性既能示踪现代海洋的循环过程,比如计算海水中的沉积通量、循环速率等,也可以在长时间尺度上提供古海洋的生产力等信息[1]。Cochran等[2]利用Greenland大陆架水体中234Th/238U亏损,计算了溶解营养盐的清除速率和颗粒有机碳通量。通过对Bahamas陆架斜坡沉积物、珊瑚和海水中234U/238U比值的分析,Henderson[3]得出过去80万年陆地上物理风化与化学风化的比例不会高于现在的结论。由此,认识铀系核素在海水中的分布和行为特征是这些核素示踪海洋过程必不可少的基础工作。
许多研究表明现代海洋中铀的收支可以用稳态箱式模型来描述:河流携带的陆源风化铀是海洋中铀的主要来源;处于低氧或缺氧环境的沉积物,生物壳体碳酸盐,热液蚀变和海床风化是海洋铀重要的汇[4]。然而,位于河流与海洋的交汇处的河口,由于具有复杂多样的生物地球化学环境,能够改变河流铀的入海通量[5]。如在亚马孙河水体输入其陆架过程中,水体环境由富氧转化为低氧,导致悬浮颗粒物中的铁锰氧化物组分被还原并且溶解,同时将结合的铀释放进入水体,该过程产生的铀添加通量可以达到亚马孙河溶解铀入海通量的5倍[6]。与此相反,在Ganges-Brahmaputra河口,由于红树林沉积物提供的还原环境造成铀由溶解态的六价转化为不可溶的四价,从而使溶解铀在整个盐度梯度上均呈现清除行为[7]。
随着电感耦合等离子体质谱仪(ICP-MS)测量技术的发展和应用,近年来铀同位素比值的测量精度有了极大地提高,这使得234U/238U可以方便地用于示踪河口水体中铀的源/汇转换和物质输运等过程[8]。例如,喜马拉雅山脉不同地貌结构单元的河流都具有各自特征的234U/238U,可指示Ganges-Brahmaputra水系中铀的来源[9];Church等[10]通过分析沉积物中腐殖酸和金属氧化物组分的234U/238U,证实了微生物捕捉和絮凝物吸附是造成Delaware盐沼潮汐水中铀清除的主要因素;在Fly河口,随着盐度升高,234U/238U显示了非线性的系统下降,但溶解铀却呈现保守行为,说明在沉积物-水界面上同时存在清除和释放两个反向过程,即溶解态和颗粒态铀在河口发生了相互交换[11]。
长江是世界上最大的河流之一,输送了大量的陆源物质进入东海(径流量8.3×1011m3/a,泥沙通量1.2×108t/a;中国河流泥沙公报2011—2014)。我们之前的工作已报道长江流域风化产物为长江口提供了(2.33±0.10)×106mol/a的溶解铀[12],大约占全球河流入海铀通量的10%。在此工作基础上,本文拟研究长江口溶解铀浓度、表层沉积物中各组分结合的铀含量和相应234U/238U比值在长江口的空间分布,长江口地球化学环境对铀迁移过程的控制机理和对铀入海通量的影响,其研究结果将为天然铀同位素及其衰变系的子体核素在海洋环境过程的示踪研究提供基础数据。
2 研究区域和实验方法
2.1 长江口概况
长江口属于强径流(年平均径流量为29 400 m3/s)、中等潮差(平均潮差2.65 m)型河口。由于流域来沙的堆积和细颗粒泥沙絮凝作用,长江口形成最大浑浊带,进而形成了拦门沙。拦门沙的纵向延伸范围约25~46 km(121.83°~122.50°E,图1)。其表层水体中悬浮颗粒物浓度在0.1~0.7 g/L之间,底层水在1~8 g/L之间[13];悬浮颗粒物浓度和水体盐度密切相关,底层泥沙从盐度为2的等值线附近开始增长,在8~15之间达到峰值[13—14]。本研究的样品采集于枯季(2014年1月和2013年3月);在此期间,东海的高盐度水体入侵进入长江口北支上段,有时会溢出进入长江口南支形成盐水倒灌现象[15]。自淡水端元(徐六泾)到最大浑浊带向海边界,河口水体的0~28.7盐度等值线密集的压缩在此范围内。
2.2 样品采集
2014年1月和2013年3月分别采集了9个表层水样和5个表层沉积物样;两次采样的区域均覆盖了从徐六泾到拦门沙向海的边界(图1)。徐六泾站位(盐度为0)作为淡水端元;千米水深站CJ的表层水样品(盐度34.4)作为海水端元(表1)。

图1 长江口采样站位图Fig.1 Sampling stations in the Changjiang Estuary表层水体(+),表层沉积物(●),长江口淡水端元——徐六泾(□),海水端元——CJ站位(▲) surface water (+); surface sediment (●); freshwater end member station-Xuliujing (XLJ/□) and seawater end member (CJ, ▲)
水样经孔径0.45 μm的醋酸纤维膜过滤;空白滤膜预先经过酸洗、烘干和称重。滤液中加入二次蒸馏浓硝酸酸化至pH值1~2之间保存待用。收集滤膜,烘干后减去空白滤膜重,得到悬浮颗粒物浓度[16]。长江口表层沉积物样品(0~1 cm)使用自制的箱式采样器采集,封入袋中冷冻保存。
表1 2014年1月长江口和东海表层水中盐度、悬浮颗粒物浓度、溶解铀浓度、过剩铀浓度、234U/238U活度比值和磷酸根浓度


Changjiang Estuary and the East China Sea during January 2014
注:NM表示没有测量。
2.3 表层沉积物序列提取实验
本文采用Tessier等[17]设计的流程进行表层沉积物的序列提取实验。实验按照以下次序提取各个组分中的铀:(1)解吸铀,(2)碳酸钙结合铀,(3)铁锰氧化物结合铀,(4)有机质结合铀。为观察颗粒态铀在河口水环境下的实际解吸情况,本研究对解吸步骤略微改动:将人工海水稀释至与各表层沉积物站位水体的实测盐度(表2)一致,所得的稀释剂用来代替Tessier等[17]原文中的MgCl2试剂;同时分析提取液中钡和钙离子。另外,使用25% HF-35% HClO4-40%HNO3混合液对表层沉积物样品进行消化[18],从而得到表层沉积物的总铀含量。总颗粒态铀含量减去序列提取实验所得的各形态铀含量的差值就是矿物晶体结构中的铀含量。
2.4 样品预处理与测
溶解铀浓度:使用0.1 mol/L稀硝酸将水体样品稀释至盐度低于0.35,加入已知量的U-236(Cat. No.7336,An Eckert & Ziegler Company)做内标,在ICP-MS(Element 2TM)上进行U浓度分析[12]。样品测量的相对标准偏差低于1.2%。在制备的U(U3O8,GB 5750-85)标准溶液中加入U-236标准溶液,定期测试流程的重现性和可靠性(频率为10%~20%)。实验全流程本底低于0.1 pmol/L。
234U/238U比值:使用磷酸钙共沉淀对水样中溶解铀进行富集后,用8 mol/L的盐酸溶解共沉淀;所得的溶液通过阴离子交换树脂(Dowex®1×8-200)纯化[12]。最终的洗脱液(基质为0.1 mol/L硝酸)在美国韦恩大学的ICP-MS(Agilent 7700)上进行测量。样品测量的相对标准偏差小于1.0%。
溶解钡浓度的分析采用SPEX CLMS-2多元素混标液做外标,同样在ICP-MS(Element 2TM)上完成测试;溶解钙浓度测试利用Ca元素标液SIMT GBW(E)做外标,在原子吸收光谱(PE 800)上测量;水样的磷酸根浓度的分析在SKALAR营养盐自动分析仪(Segmented Flow Analyzer,SanplusSystem)上进行[12]。
粒度分析使用镭射粒径分析仪(Beckman coulter®LS 13 320)进行测量;盐度分析使用盐度计SYA 2-2完成。
3 结果
3.1 长江口铀的空间分布特征
长江口表层水中溶解铀浓度,234U/238U活度比值列于表1。长江口淡水端元溶解铀的浓度为 (4.22±0.05)nmol/L,与我们之前报道的浓度范围(1.32~4.06 nmol/L)相比略高[12]。由于采样时间属于长江的枯季,这个结果符合“长江的径流量对溶解铀浓度有稀释作用,因此枯季铀浓度处于较高水平”的结论[12]。海水端元的溶解铀浓度和234U/238U活度比值分别为(13.12±0.09) nmol/L和(1.145±0.001),与Chen等[19]早期报道的全球海洋平均值13.5 nmol/L和1.148(盐度35)相近。因此,在溶解铀-盐度关系图上直线连接淡水和海水端元点就可以得到如图2所示的溶解铀的保守混合线。样品的实测值减去保守值就可得到过剩铀浓度(ΔU,nmol/L)。另外,利用我们先前报道的长江口淡水端元234U/238U枯季值1.424[12],溶解态234U/238U活度比值的保守混合值可表达为[20]:

(1)
式中,A表示234U和238U同位素的活度,S表示盐度。下标RW、SW分别代表河水和海水。公式(1)随着河口盐度增长而产生的理想保守混合曲线如图2所示。

图2 溶解铀浓度(□)、水体234U/238U活度比值(◇)、悬浮颗粒物浓度(○)和表层沉积物中颗粒态可释放铀的234U/238U活度比值与水体盐度关系图:直线表示溶解铀保守混合线。水体234U/238U活度比值的保守混合曲线由文中公式(1)定义Fig.2 DUC (□), dissolved 234U/238U (◇), SPM concentration (○) and the 234U/238U AR of releasable particulate U (△) and salinity in water column. The solid line/curve represented the DUC/234U/238U AR and salinity relationship for ideal mixing of river water and seawater. The conservative mixing curve of the dis-solved 234U/238U AR was defined by eq. (1) in text
从图2可以看出,低盐度区(南支)的实测溶解铀浓度与保守值一致,但234U/238U比值低于保守值;相反,中高盐度区(北支和拦门沙)呈现明显的溶解铀富集现象,而234U/238U与保守值基本一致(图2)。过剩铀在整个调查区域的空间分布情况如图3所示:在北支和拦门沙地区,伴随着盐度由3.7(K1306)增长至23.8(K1301),ΔU由0.67 nmol/L增至2.74 nmol/L(表1);位于拦门沙向海边界的K1315和K1313站位,虽然盐度分别增加到25.6和27.8,但ΔU分别为2.34、2.07 nmol/L,呈下降趋势,与之前变化趋势相反。

图3 长江口铀添加和清除程度的空间分布Fig.3 Spatial distribution patterns of U addition and depletion extents in the Changjiang Estuary饼状图表示过剩铀(深灰色扇形)或清除铀(白色扇形)占保守铀(整个圆形)的比例。等值线为采样期间的盐度分布 The pie chart showed the proportion between the excess U(dark gray sector)/depleted U (white sector) and the conservative U(the entire circle). The contour lines showed salinity dis-tribution during sampling period
3.2 序列提取实验结果
表2展示了长江口表层沉积物中各个组分(解吸、碳酸钙、铁锰氧化物、有机物和总颗粒态)结合铀的含量和234U/238U比值。O1、O8、A6-2和A6-5站位是细颗粒物样品(中值粒径范围在8.21~17.62 μm之间);O4是一个粗砂样品站位(中值粒径177.1 μm)。细颗粒物样品的总铀含量非常接近,平均值为(12.38±0.01)μmol/kg。粗砂样品的总铀含量为(5.39±0.04) μmol/kg。细颗粒样品的可解吸铀比例从河口淡水端元(O8)的0.2%增长至拦门沙地区(A6-2)的4.1%;而在更加向海的A6-5站位,可解吸铀含量下降至2.1%(表2)。与此同时,可解吸钡含量表现出与铀一致的空间分布[O8站位:(0.32±0.03) μmol/kg;O1站位:(86.42±0.60) μmol/kg;A6-2站位:(103.0±2.0) μmol/kg;A6-5站位:(8.87±0.21) μmol/kg)]。实验前后提取液中钙离子浓度变化量分别为0.27 mmol/L(O8站位),0.36 mmol/L(O4站位),-0.21 mmol/L(O1站位),-1.58 mmol/L(A6-2站位)和-5.99 mmol/L (A6-5站位)。
碳酸钙结合铀含量占总颗粒铀含量的比例从淡水端元的13.0%(O8)急剧下降至拦门沙地区的0.7%(A6-2站位)。铁锰氧化物和有机物结合铀的含量相对稳定,占总颗粒态铀含量的比例分别在2.4%~4.0%和4.0%~4.8%之间。另外,与细颗粒物相比,粗砂的解吸铀(0.1%)和铁锰氧化物结合铀(1.8%)的比例明显偏低。
4 讨论
4.1 泥沙中颗粒态铀释放对河口溶解铀的影响
枯季东海盐水入侵长江口北支,导致北支上段的盐度增长至20以上。图3显示北支和拦门沙地区的水体中都观测到了铀富集的现象。已有文献报道海底地下水排放和颗粒态铀释放都有可能造成水体中铀富集[6,9]。由于地下水往往具有高234U/238U比值,其排放会造成水体中铀同位素比值的升高[4],这明显与我们观察到的现象不一致(图2)。但进一步分析表明,如果不考虑K1315和K1313站位(拦门沙的向海边界),ΔU和悬浮颗粒物浓度(SPM)之间有非常好的相关性 (r2=0.96,图4),说明悬浮颗粒物很可能是水体中过剩铀的重要来源。

图4 过剩铀与悬浮颗粒物浓度线性拟合关系图)拟合不包括拦门沙向海边界站位K1313和K1315)Fig.4 Linear fitting plots of the excess U and SPM:the fitting points excluded the stations K1313 and K1315
长江口的悬浮颗粒物包括两个部分,由径流输送的泥沙和河口表层沉积物再悬浮。其中,河流泥沙的输运最远能到达123°E以西的位置[21]。而在122.5°E附近(拦门沙向海边界),由于长江径流的水动力减弱,导致流域来沙输运过程减缓,悬浮(包括再悬浮)颗粒物浓度降低,水体由混浊向清澈转变[22]。另外,长江口表层沉积物序列提取实验显示铀与钡同时发生解吸,并且空间变化趋势一致,即从淡水端元增长至拦门沙地区达到最高值,之后在拦门沙向海边界处下降,说明颗粒物在经过拦门沙地区的输运过程中,可解吸铀和钡几乎完全释放。上述两个因素——拦门沙地区的高悬浮颗粒物浓度和高铀解吸量能够为这个区域贡献高的过剩铀浓度。然而,在拦门沙向海边界,悬浮颗粒物浓度降低并且铀释放过程变弱导致过剩铀浓度降低。
碳酸钙结合铀的空间分布显示,从淡水端元到拦门沙地区,碳酸钙组分损失了大约1.51 μmol/kg铀含量(表2)。事实上,解吸实验完成后,低盐度区O8和O4站位的提取液中钙浓度增加,说明颗粒物中钙溶出;但在高盐度区O1、A6-2和A6-5站位,提取液中钙浓度又有减小,说明水体中更多的钙可能通过离子交换进入沉积物。Loire河口也出现了沉积物输运过程中颗粒态钙含量降低的现象[23],Abril等[24]提出这是由于在富营养化条件下发生了碳酸钙的溶解。在黄河流域也观察到了相似的现象,黄土中的碳酸钙结合铀可释放进入水体[25]。由此推断,碳酸钙结合铀可能是长江口过剩铀的另一个主要来源。
如前所述,在亚马孙陆架水体中观测到十分明显的溶解态铀过剩现象:ΔU最大值为11.81 nmol/L,为长江口的4倍之多;由于氧化还原环境的变化使颗粒物中铁锰氧化物组分结合铀释放是亚马孙陆架水体中过剩铀的主要来源[6]。然而,长江口细颗粒物中铁锰氧化物结合铀含量变化非常小(淡水端元和拦门沙地区的差值仅占颗粒态总铀含量的1.4%,表2),远低于亚马孙陆架的44.0%[6]。因此,长江口铁锰氧化物结合铀应该不是过剩铀的主要来源。另外,粗砂仅占长江泥沙排放总量的6%,在长江口表层沉积物中的分布也非常少;并且,与亚马孙陆架相似,长江口粗砂样品中的铀含量非常低,因此长江口颗粒悬浮物或沉积物中粗砂对水体中过剩铀的贡献是可以忽略的[26—27]。
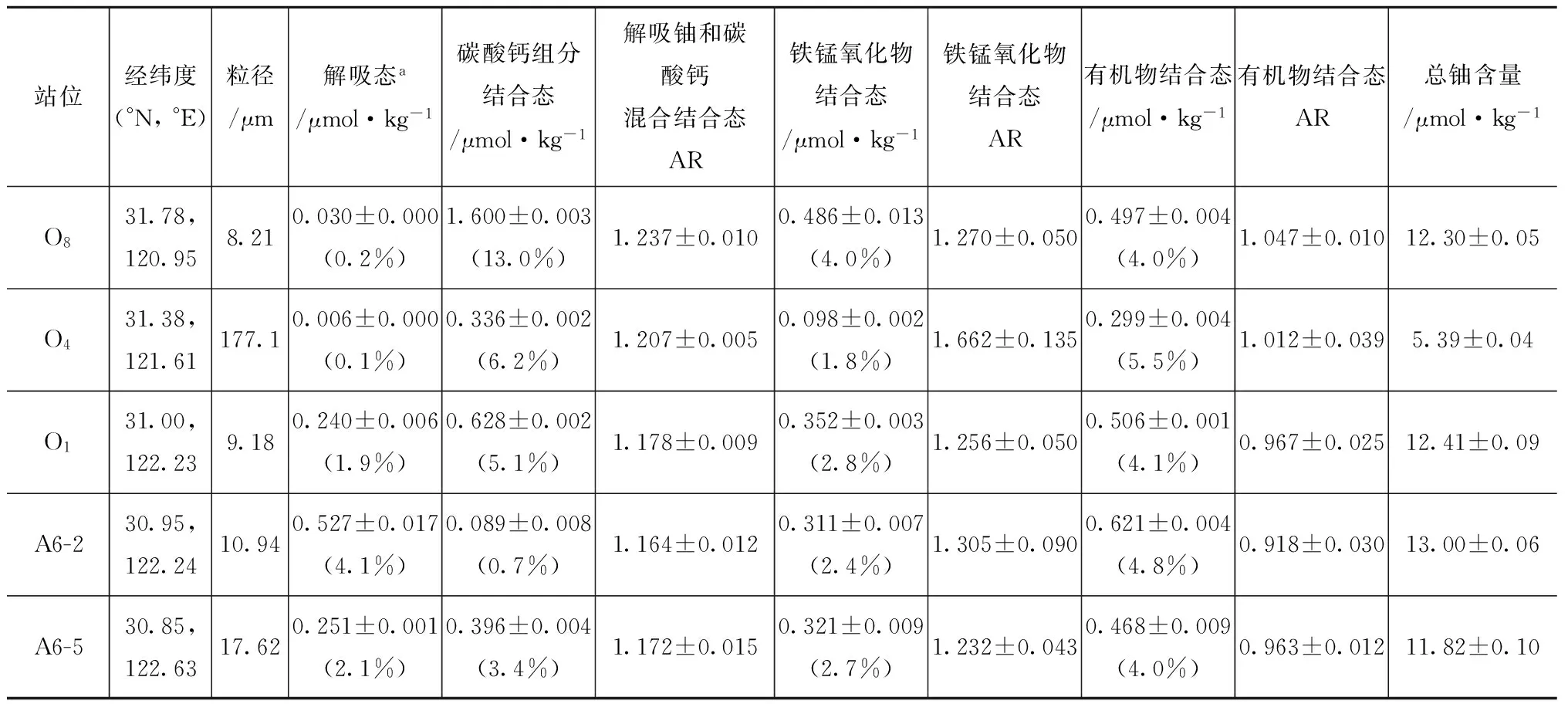
表2 长江口表层沉积物中各矿物组分结合态的铀含量(μmol/kg)和234U/238U活度比值(AR)
注:a解吸铀组分是由与表层沉积物样品采样站位水体盐度一致的人工海水稀释剂进行淋滤的 (O8站位盐度0.2,O4站位盐度0.4,O1站位盐度15.7,A6-2站位盐度18.2,A6-5站位盐度23)。%表示各组分结合铀占总铀含量的百分比。
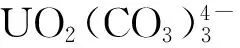
4.2 铀的“伪保守”行为
两个低盐度站位(K1304和K1305)的实测溶解铀浓度接近保守值,但其234U/238U活度比值却处于保守线的下方(图2),说明除了长江径流和外海水的贡献,还有其他铀的来源,并且这种来源应该具有更低的234U/238U活度比值。类似的现象在Zaire河口也有报道[31]。
将每个表层沉积物样品的解吸组分和碳酸钙组分的提取液混合后进行234U/238U比值的测量,发现O8和O4站位的活度比值分别为1.237和1.207(表2)。图2表明,在低盐度的长江口南支,颗粒态可释放铀的234U/238U活度比值要低于实测水体值,而实测水体值又低于保守值;这进一步证明颗粒物态铀释放进入长江口水体成为具有低比值的铀源,导致水体中的铀活度比值降低,是水体中过剩铀的主要来源。
除了上述过程,还有一些过程能够引起铀的清除。比如在长江口低盐度区,铁氢氧化物形成絮凝造成铁离子从水体中清除[32—33];在这个过程中,溶解态的六价铀能够被絮凝物吸附,与絮凝物一同沉降之后被埋藏进入沉积物[10,34]。另外,长江口最大浑浊带颗粒物再悬浮和聚集絮凝作用活跃,也可能造成铀的清除[13]。
颗粒态铀释放过程与溶解铀清除过程发生,导致铀在颗粒相和水相中动态交换频繁。在其交换过程中,水体中溶解铀和颗粒态可释放铀的活度比值随着长江口水体盐度升高逐渐接近保守混合曲线(图2)。因此,低盐度区溶解铀表观上的“保守混合”,并不是简单的河水和海水的双端元混合过程,而是当添加项和清除项的发生速率相近呈现出的“伪保守”行为。类似的溶解态和颗粒态铀交换在Kalix河口也有报道[35]。与长江口类似,Swarzenski等[11]的工作也表明,当Fly河水流入河口三角洲后,铀同位素在水体—胶体—颗粒物三态之间重新分配导致低盐度区的表层水体234U/238U活度比值发生变化,最终在中-高盐度区域达到新的平衡。
4.3 长江口颗粒物对溶解铀的添加通量
4.3.1 溶解态和颗粒态铀的河流输入通量
我们先前的研究工作已表明由长江径流输入河口的溶解铀通量为(2.33±0.10)×106mol/a,占全球河流溶解铀通量的10.1%[12]。另外,长江流域的上游奉节,中游武汉和下游大通的土壤样品中的铀含量分别为(13.68±0.17)、(21.93±0.42)和(9.67±0.11) μmol/kg,平均值为(15.09±0.90) μmol/kg[12];这个值与长江口淡水端元(O8站位)表层沉积物的铀含量[(12.23±0.05) μmol/kg,表2]相近。由于河流泥沙主要来自于流域的物理风化,因此若利用流域土壤的铀含量平均值代表长江输运的颗粒态铀含量与平均泥沙通量(119×106t/a,中国河流泥沙公报2011-2014)之乘积,即可得到颗粒态铀通量为(1.80±0.17)×106mol/a;这个值是输入长江口总铀通量(溶解态加颗粒态之和)的43%,构成全球河流颗粒态铀通量(250×106mol/a)的0.72%[3]。
4.3.2 颗粒态铀的释放通量
根据ΔU与SPM之间非常强的正相关性(r2=0.96,图4),可估算出颗粒态铀的释放通量:将大通水文站的平均SPM(0.14 g/L,长江泥沙入海年通量除以径流年通量,中国河流泥沙公报2011-2014)代入ΔU和SPM的拟合公式ΔU =4.36×SPM-0.20,得到水体中的ΔU的平均值,为(0.41±0.03)nmol/L;利用此值乘以长江年径流量,即可得到颗粒态铀的释放通量为(3.48±0.41)×105mol/a。这意味着19.3%的河流颗粒态铀在长江口可以转化为溶解铀进入水体。
5 结论
本论文通过采集长江口表层水体和表层沉积物样品,测量溶解铀浓度,颗粒态铀含量和相应的234U/238U比值,研究长江口铀的地球化学行为及其影响因素,主要结论如下:
(1)长江口呈现出明显的溶解铀富集现象。悬浮颗粒物浓度和水体中过剩铀之间有显著相关性(r2=0.96),说明颗粒态铀释放应该是过剩铀的重要来源。
(2)表层沉积物的序列提取实验说明每千克颗粒物或沉积物能通过解吸和碳酸钙组分释放约2 μmol颗粒态铀进入长江口水体,占总颗粒态铀含量(15.09 μmol/kg)的13.3%。
(3)在长江口,颗粒态铀释放过程和溶解铀清除过程同时发生使铀在水相和颗粒相之间重新分配。在低盐度区域,颗粒物释放低234U/238U比值的铀混合进入水体,使得水体234U/238U比值低于保守值;但若铀的添加速率和清除速率相近,溶解铀与盐度的关系就会呈现出表观上的“保守”行为。而在中高盐度区,溶解铀富集,但溶解铀与颗粒态可释放铀的234U/238U活度比值接近说明活跃的铀交换过程使铀在水相和颗粒相之间的分配达到新的平衡。
(4)长江口颗粒态铀释放通量为(3.48±0.41)×105mol/a,占长江总颗粒态铀通量(1.80±0.17)×106mol/a的19.3%。由长江口输入东海的溶解铀总通量(河流溶解铀与河口添加铀之和)为(2.68±0.13)×106mol/a,约为世界河流入海铀通量的11.7%。
致谢:衷心感谢美国韦恩州立大学人文科学学院地质系Mark Baskaran教授实验室协助测量铀同位素活度比值样品;感谢华东师范大学河口海岸学国家重点实验室张卫国老师实验室提供长江口表层水体样品的帮助。
[1]Henderson G M,Anderson R F. The U-series toolbox for paleoceanography[J]. Reviews in Mineralogy and Geochemistry,2003,52(1):493-531,doi:10.2113/0520493.
[2]Cochran J K,Barnes C,Achman D,et al. Thorium-234/Uranium-238 disequilibrium as an indicator of scavenging rates and participate organic carbon fluxes in the Northeast Water Polynya,Greenland[J]. Journal of geophysical research,1995,100(C3):4399-4410.
[3]Henderson G M.Seawater (234U/238U) during the last 800 thousand years[J]. Earth and Planetary Science Letters,2002,199(1/2):97-110
[4] Dunk R,Mills R,Jenkins W. A reevaluation of the oceanic uranium budget for the Holocene[J]. Chemical Geology,2002,190(1/4):45-67
[5] McKee B A. Chapter 6 U-and Th-Series nuclides in estuarine environments[J]. Radioactivity in the Environment,2008,13:193-225
[6] McKee B A,DeMaster D J,Nittrouer C A. Uranium geochemistry on the Amazon Shelf:Evidence for uranium release from bottom sediments[J]. Geochimica et Cosmochimica Acta,1987,51(10):2779-2786.
[7] Carroll J,Moore W S. Uranium removal during low discharge in the Ganges-Brahmaputra mixing zone[J]. Geochimica et Cosmochimica Acta,1993,57(21/22):4987-4995.
[8]Chabaux F,Riotte J,Deauincev O. U-Th-Ra fractionation during weathering and river transport[J]. Reviews in Mineralogy and Geochemistry,2003,52(1):533-576.
[9]Chabaux F,Riotte J,Clauer N,et al. Isotopic tracing of the dissolved U fluxes of Himalayan Rivers:Implications for present and past U budgets of the Ganges-Brahmaputra system[J]. Geochimica et Cosmochimica Acta,2001,65(19):3201-3217.
[10]Church T M,Sarin M M,Fleisher M Q,et al. Salt marshes:An important coastal sink for dissolved uranium[J]. Geochimica et Cosmochimica Acta,1996,60(20):3879-3887.
[11] Swarzenski P W Campbell P,Porcelli D,et al. The estuarine chemistry and isotope systematics of234,238U in the Amazon and Fly Rivers[J]. Continental Shelf Research,2004,24(19):2357-2372
[12] Zhou Jing,Du Jinzhou,MooreW S,et al. Concentrations and fluxes of uranium in two major Chinese rivers:The Changjiang River and the Huanghe River[J].Estuarine,Coastal and Shelf Science,2015,152:56-64.
[13]沈焕庭,贺松林,潘定安,等.长江河口最大浑浊带研究[J]. 地理学报,1992,47(5):472-479.
Shen Huanting,He Songlin,Pan Ding’an,et al.A study of turbidity maximum in the Changjiang Estuary[J]. Acta Geographica Sinica,1992,47(5):472-479.
[14] 陈沈良,胡方西,胡辉,等.长江口区河海划界自然条件及方案探讨[J]. 海洋学研究,2009,27(S1):1-9.
Chen Shenliang,Hu Fangxi,Hu Hui,et al. On the natural conditions and program for river-sea delimitation in Changjiang River Estuary[J]. Journal of Marine Sciences,2009,27(S1):1-9.
[15]Wu Hui,Zhu Jianrong,Chen Bingrui,et al. Quantitative relationship of runoff and tide to saltwater spilling over from the North Branch in the Changjiang Estuary:a numerical study[J]. Estuarine,Coastal and Shelf Science,2006,69(1/2):125-132
[16] Li Chao,Yang Shouye,Zhang Weiguo. Magnetic properties of sediments from major rivers,aeolian dust,loess soil and desert in China[J]. Journal of Asian Earth Sciences,2012,45:190-200.
[17] Tessier A,Campbell P G C,Bisson M. Sequential extraction procedure for the speciation of particulate trace metals[J]. Analytical Chemistry,1979,51(7):844-851.
[19]Chen J H,Edwards R L,Wasserburg G J.238U,234U and232Th in seawater[J]. Earth and Planetary Science Letters,1986,80(3/4):241-251.
[20] Toole J,Baxter M S,Thomson J. The behavior of uranium isotopes with salinity change in three U. K. estuaries[J]. Estuarine,Coastal and Shelf Science,1987,25(3):283-297
[21] 史立人,魏特,沈蕙漱.长江入海泥沙扩散与北支淤积泥沙来源[J]. 长江水利水电科学研究院院报,1985(2):8-19.
Shi Liren,Wei Te,Shen Huishu. Seaward sediments from Yangtze River and the source of depositing sediments in the North Branch[J]. Journal of Yangtze River Scientific Research Institute,1985(2):8-19.
[22] Shi J Z. Tidal resuspension and transport processes of fine sediment within the river plume in the partially-mixed Changjiang River estuary,China:A personal perspective[J]. Geomorphology ,2010 ,121(3/4):133-151.
[23] Negrel P. Multi-element chemistry of Loire estuary sediments:anthropogenicvs. natural sources[J]. Estuarine,Coastal and Shelf Science,1997,44(4):395-410.
[24] Abril G,Etcheber H,Delille B,et al. Carbonate dissolution in the turbid and eutrophic Loire estuary[J]. Marine Ecology. Progress Series,2003,259:129-138.
[25] Jiang Xueyan,Yu Zhigang,Ku T L,et al. Distribution of uranium isotopes in the main channel of Yellow River (Huanghe),China[J]. Continental Shelf Research,2009,29(4):719-727.
[26] Li Chen,Wu Menwu,Zhang Junyong. Effect of the three gorges project on sediment transportation of the Yangtze estuary[J]. Resources and Environment in the Yangtze Basin,2003,12(1):50-54.
[27] Lin S,Hsieh I J,Huang Kuoming,et al. Influence of the Yangtze River and grain size on the spatial variations of heavy metals and organic carbon in the East China Sea continental shelf sediments[J]. Chemical Geology,2002,182(2/4):377-394.
[28] Zhai Weidong,Dai Minhan,Guo Xianghui. Carbonate system and CO2degassing fluxes in the inner estuary of Changjiang (Yangtze) River,China[J]. Marine Chemistry,2007,107(3):342-356,doi:10.1016/j.marchem.2007.02.011.
[29] Dang Hongyue,Zhang Xiaoxia,Sun Jin,et al. Diversity and spatial distribution of sediment ammonia-oxidizing crenarchaeota in response to estuarine and environmental gradients in the Changjiang Estuary and East China Sea[J]. Microbiology,2008,154:2084-2095
[30] Sarin M M,Church T M. Behaviour of uranium during mixing in the Delaware and Chesapeake Estuaries[J]. Estuarine,Coastal and Shelf Science,1994,39(6):619-631.
[31] Martin J M,Meybeck M,Pusset M. Uranium behaviour in the Zaire estuary[J]. Netherlands Journal of Sea Research,1978,12(3/4):338-344.
[32] Edmond J M,Spivack A,Grant B,et al. Chemical dynamics of the Changjiang Estuary[J]. Continental Shelf Research,1985,4(1/2):17-36.
[33] Bergquist B A,Boyle E A. Iron isotopes in the Amazon River system:Weathering and transport signatures[J]. Earth and Planetary Science Letters,2006,248(1/2):54-68.
[34] Swarzenski P W,Mckee B A,Booth J G. Uranium geochemistry on the Amazon Shelf:Chemical phase partitioning and cycling across a salinity gradient[J]. Geochimica et Cosmochimica Acta,1995,59(1):7-18.
[35] Andersson P S,Porcelli D,Gustafsson O,et al. The importance of colloids for the behavior of uranium isotopes in the low-salinity zone of a stable estuary[J].Geochimica et Cosmochimica Acta,2001,65(1):13-25.
The importance of the suspended sediment for the uranium non-conservative behavior in the Changjiang Estuary
Zhou Jing1,Du Jinzhou1,Bi Qianqian1,Wang Jinlong1,Liu Dantong1
(1.StateKeyLaboratoryofEstuarineandCoastalResearch,EastChinaNormalUniversity,Shanghai200062,China)
To study geochemical behavior of uranium in the Changjiang Estuary (CJE),surface water and surface sediment samples were collected along a salinity gradient during January 2014 and March 2013, respectively. The dissolved uranium concentration (DUC) and234U/238U activity ratio (AR) were measured by the inductively coupled plasma mass spectrometer (ICP-MS). Sequential extraction experiments of surface sediments (collected from the freshwater end-member to the seaward boundary of the sandbar) were also conducted to analyze spatial distribution of U components and234U/238U ARs in four fractions:desorption,calcium carbonate,Fe-Mn oxide and organic matter. The results showed that there is an extra source of the dissolved uranium into the CJE by other processes excepting U supplied from the ideal mixing process of river water and seawater. The excess DUC and suspended particle matter (SPM) showed a strong positive correlation (r2=0.96). Around 2 μmol U can be transferred from per kilogram particulate into aqueous phase through the desorption and calcium carbonate dissolution,but this phenomenon did not occur for the Fe-Mn oxides and particulate organic matter. At the low salinity region,the releasable particulate U with low234U/238U ARs resulted in the measured234U/238U ARs of the water column below the conservative mixing curve,although the DUC points fell on the conservative mixing line. However,flocculations of the iron hydroxide and fine particle may cause simultaneous depletion of the dissolved uranium. Consequently,when the U input and removal rates were approximately equal at the low salinity region,the dissolved uranium showed apparently conservative phenomenon. At the mid-high salinity region (with high SPM),excess dissolved uranium was observed in the water column,but the U exchange between the particulate and aqueous phases led to that the releasable234U/238U ARs of the particles were close to the measured234U/238U ARs in the water column.The excess dissolved uranium flux from the particle was estimated as (3.48±0.41)×105mol/a which occupied 19.3% particlulate U flux [(1.80±0.17)×106mol/a] into the CJE. The total flux (i.e.,riverine flux plus additional flux) of the dissolved uranium from the CJE [about (2.68±0.13)×106mol/a] contributed about 11.7% of the DU input to the world ocean.
dissolved uranium;particulate uranium;234U/238U AR;non-conservation;Changjiang Estuary
2016-06-14;
2016-09-30。
国家自然科学基金(41240038,41376089)。
周婧(1984—),甘肃省兰州市人,主要研究方向为近海同位素生物地球化学。E-mail:jingchow@outlook.com
*通信作者:杜金洲,教授,主要研究方向为同位素海洋学,环境放射化学,近海生物地球化学过程。E-mail:jzdu@sklec.ecnu.edu.cn
10.3969/j.issn.0253-4193.2016.12.005
P734
A
0253-4193(2016)12-0046-09
周婧,杜金洲,毕倩倩,等. 悬浮泥沙在长江口铀非保守行为中的重要作用[J].海洋学报,2016,38(12):46—54,
Zhou Jing,Du Jinzhou,Bi Qianqian,et al. The importance of the suspended sediment for the uranium non-conservative behavior in the Changjiang Estuary[J]. Haiyang Xuebao,2016,38(12):46—54,doi:10.3969/j.issn.0253-4193.2016.12.005
