总有机碳精密度偏性试验数据分析
2016-12-15谢卫民卢云黎马碧文
谢卫民 卢云黎 马碧文
(长江委水文局长江中游水环境监测中心,湖北 武汉 430012)
总有机碳精密度偏性试验数据分析
谢卫民 卢云黎 马碧文
(长江委水文局长江中游水环境监测中心,湖北 武汉 430012)
依靠精密度偏性试验对实验室总有机碳(TOC)分析进行控制检查,利用实验室空白检验、标准偏差检验、加标回收率检验等方法全面评价总有机碳试验数据的精密度和准确度;并根据试验数据绘制质量控制图,检查试验受控状态。根据试验数据得知,实验室实际检测限低于分析方法检测限,批内、批间变异结果为“不显著”,加标回收率也在可接受范围内,质控图中质控点偏离均在可控范围内。依据分析方法检测时,实验室人员、仪器设备及试验环境均可以保证TOC的检测精准度,检测结果可靠。
污染;检测限;精密度偏性试验;总有机碳 ;质控
总有机碳(TOC)是指溶解或悬浮在水中有机物的含碳量,是以含碳量表示水体中有机物总量的综合指标,它直接反映了水体被有机物污染的程度。
实验室依靠精密度偏性试验和质量控制图技术,确定实验室总有机碳测定的精密度和准确度,使用质量控制图、加标回收率控制、空白试验、标准偏差检验等方法,对影响分析测定的各种因素,包括试验人员、试验试剂、试验仪器、试验环境等各种可能影响测定结果的因素进行了分析,为保证实验室检测数据准确可靠提供了科学依据,为燃烧氧化-非分散红外吸收法测定总有机碳的研究提供了参考。
1 仪器及试剂
试验仪器包括:multi N/C 2100非分散红外吸收TOC分析仪、UPH-Ⅱ-20T优普超纯水制造仪、纯度大于 99.99%氧气。
试剂包括:邻苯二甲酸氢钾、碳酸氢钠、硫酸,试剂配制所用水为高纯水(无二氧化碳水)。
2 方法与结果
分析方法采用《水质-总有机碳的测定 燃烧氧化-非分散红外吸收氧化法》(HJ501—2009)中的直接法测定。在900℃~950℃下,以铂和三氧化钴或三氧化二铬为催化剂,水样中的有机物在石英管中裂解转化为CO2,使用红外线气体分析仪测定转化的CO2含量,从而计算出水样中碳的含量。
2.1 标准溶液
有机碳标准溶液(浓度500 mg/L):准确称取邻苯二甲酸氢钾(预先在110℃~120℃下干燥至恒重)0.265 6 g,置于烧杯中,加高纯水(或无二氧化碳水)溶解后,转移此溶液至250 mL容量瓶中,稀释至标线,混匀。
2.2 标准曲线
在一组6个50 mL容量瓶中,分别加入0.0,1.0,2.5,5.0,7.5mL和10.0 mL有机碳标准溶液,用水稀释至标线,混匀,配制成有机碳浓度0.0,10.0,25.0,50.0,75.0 mg/L和100.0 mg/L的标准系列浓度。将酸化至pH值小于2后的标准系列溶液注入TOC分析仪,经曝气除去无机碳后导入高温氧化炉,记录相应的仪器响应值,测定结果见表1。
以浓度值为横坐标,仪器响应值为纵坐标,绘制标准曲线。计算线性回归方程为:A=272C+46.2,r=0.999 8
结果表明,使用燃烧氧化-非分散红外吸收氧
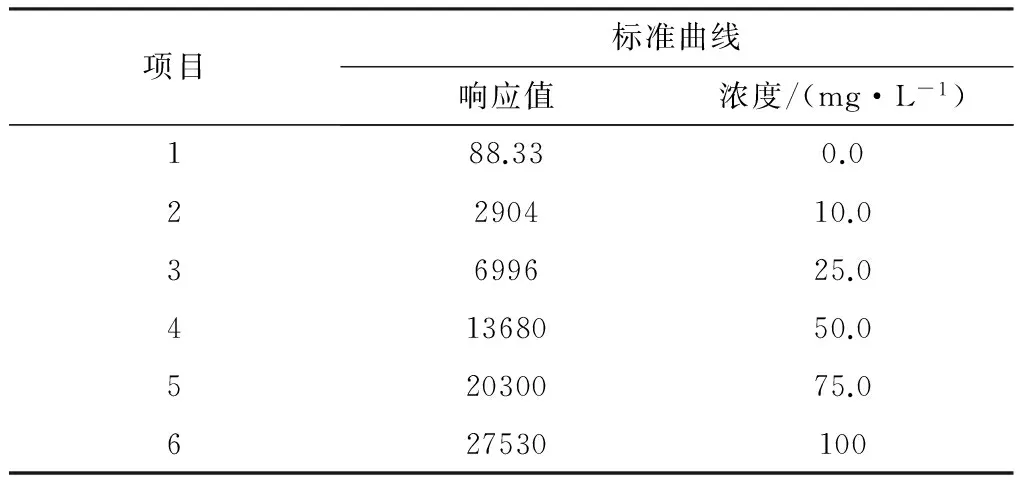
表1 标准曲线测定结果
化法和TOC分析仪时,在0.0~100 mg/L的测定范围内具有良好的线性关系,相关系数一般都能达到0.999以上。另外,对截距进行t检验,根据公式计算t=0.297,小于t0.05(n=4)=2.776,说明截距与零无显著性差异。

表2 精密度偏性试验结果
2.3 精密度试验及结果
依据《水质-总有机碳的测定 燃烧氧化-非分散红外吸收氧化法》(HJ501—2009),以随机次序测定以下6种溶液,每天测定一批,每批平行测定2份,共测定10批,测定结果见表2。
(1) 空白溶液。使用高纯水(无二氧化碳水)按样品相同方法处置。
(2) 0.1C标准溶液。10 mg/L的标准溶液。
(3) 0.9C标准溶液。90 mg/L的标准溶液。
(4) 天然水样。含有一定浓度待测物的天然水样(本次使用武汉长江干流水样)。
(5) 加标天然水样。用吸管取1.0 mL有机碳标准溶液于50 mL容量瓶中,用天然水样稀释至标线,混匀。
(6) 标准物质样品。浓度u=40.0 mg/L的标准物质,误差范围±5%。
3 结果评价分析
3.1 实验室条件下方法检出限
根据空白试验所得的测定结果,按下表中的公式计算出批内标准差及本次精密度偏性试验下的实际检出限,计算结果见表3。

表3 批内标准偏差及实际检出限
注:x为每批中单个测定值,X为每批总和,m为批数,n为每批测定次数。
由上表可知,实验室实际检出限为0.000 0mg/L。《水质-总有机碳的测定 燃烧氧化-非分散红外吸收氧化法》(HJ501—2009)中的规定检出限为0.1 mg/L,因此,实验室实际检出限小于方法检出限,说明在实验室的仪器和环境下测定TOC时空白试验符合方法标准要求。
3.2 变异因素分析
根据0.1C样品、0.9C样品、天然水样、天然加标水样和标准物质样品的测定结果,按表中的公式计算出批内变异系数和批间变异系数,同时进行变异显著性检验(即F检验)。
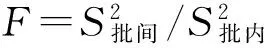

表4 批内变异、批间变异计算
可查表得知,说明变异结果为“无显著性差异”,表明实验室分析人员的操作技能稳定熟练,检测结果未受试验环境、仪器设备、试剂用水等各因素影响,试验检测结果精密度良好。
3.3 标准偏差与干扰因素分析
对0.1C样品、0.9C样品、天然水样、天然加标水样、标准物质样品的总变异系数和总标准差进行计算,计算结果见表5。
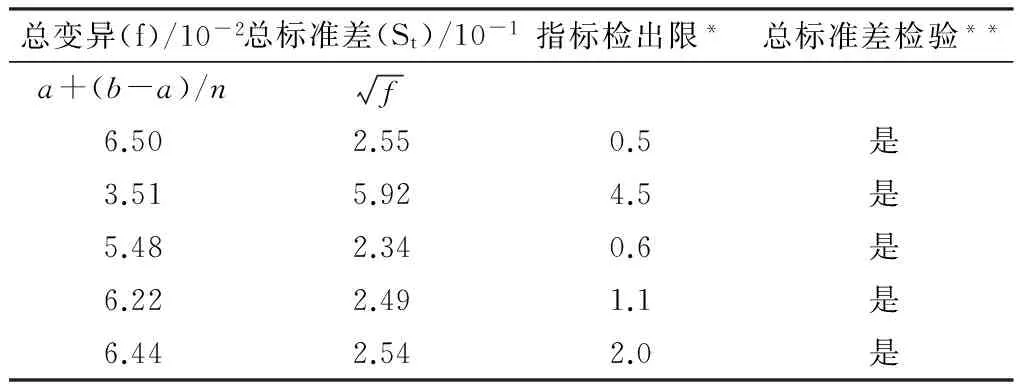
表5 总变异系数和总标准差计算结果
注:*表明两者的检出限最大值为w;**表明总标准差合格St
根据表5计算结果,总标准差均小于指标检出限,表明实验室分析环境条件和仪器设备稳定性均良好。另外,根据0.1C样品、0.9C样品、天然水样、天然加标水样的总标准差,可以确定水样中无影响检测精密度的干扰因素。
3.4 准确度分析
本次试验共测定了10批标准物质样品和天然水样、天然加标水样,分别计算标准物质样品的相对误差和天然水样加标回收率,并进行检验评价,结果见表6,7。

表6 标准物质检测相对误差计算
根据表6计算结果,标准物质真值为40.0 mg/L,标准物质测定均值为40.1 mg/L,其相对误差RE=0.23% ,小于一般要求的5%,表明TOC标准样品检测结果合格,检测过程中不存在系统误差,偶然误差在可接受的范围内。根据表7计算结果,平均加标回收率为98.4%,且所有批次加标回收率均在95%~105%,表明实验室检测结果的准确度是可靠的。
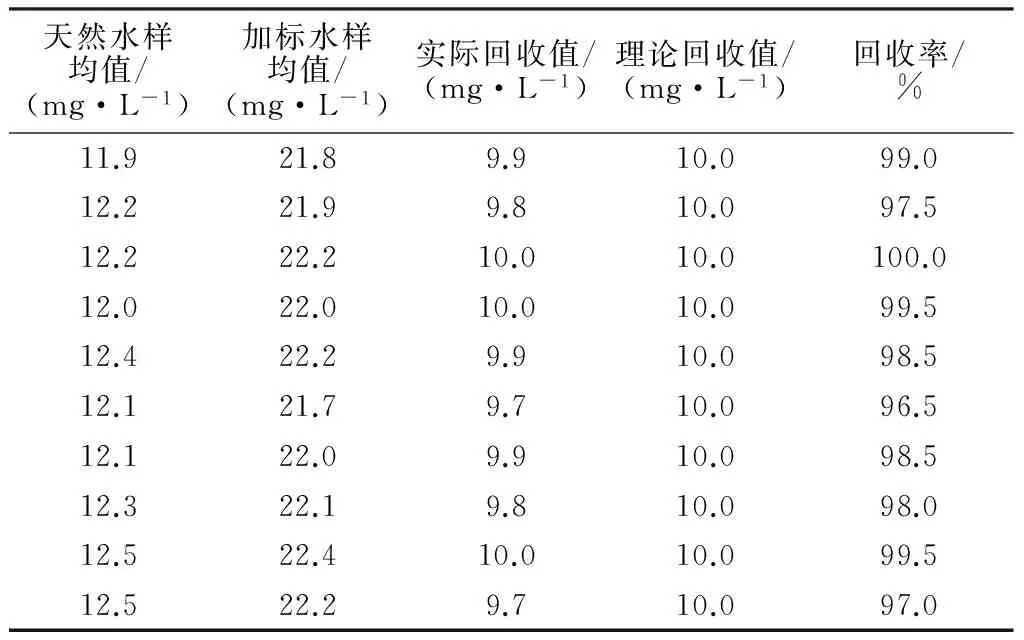
表7 天然水样加标回收率计算
3.5 质量控制图的绘制与分析
根据以上检验计算,证明实验室检测结果的精密度和准确度均符合质量控制要求,在此基础上将检测结果绘制成质量控制图,见图1。

图1 质量控制
由图2中可以看出,检测结果随机排列在中心线两侧,落入上下辅助线内的检测结果超过50%;没有连续7次检测结果出现在中心线的同一侧;也没有连续7次检测结果呈递升或递降趋势;检测结果100%均落入控制限内;没有3次检测结果中的两次连续接近控制限;无离群检测数据。以上情况说明误差呈随机状态分布在均值的两侧,检测结果没有发生系统性偏离,均在控制范围之内,是可信的。
4 结 论
通过精密度偏性试验,分析计算了实验室开展TOC参数检测时的实际检出限、检测数据的精密度、检测结果的准确度等指标,检测结果表明该参数实验室实际检出限小于分析方法规定的检出限,空白检测达到质量控制要求,实验室化学试剂、分析用水、检测环境、仪器设备均能达到方法规定要求。对标准物质样品、平行样品、加标回收样品的检测结果分别进行总体方差检验(即F检验)和总标准差检验均合格,说明检测人员的操作技能熟练,实验室环境条件和仪器设备稳定可靠,检测结果的精密度良好;根据标准物质检测和加标回收样品检测,标准物质的实际测定误差小于允许误差,加标回收率计算结果均在95%~105%的可接受范围之内,表明检测过程中的系统误差在可控范围之内,结果准确可靠。通过绘制质量控制图,进一步直观描述了检测数据的受控状态,误差呈随机状态分布在均值的两侧,检测结果没有发生系统性偏离。
本次精密度偏性试验系统地检查了检测人员技能熟练度、仪器设备稳定性、试验环境和条件的控制能力,证明了该实验室检测TOC结果的精密性和准确性,说明实验室具备同水质监测工作要求相适应的质量控制能力和检测能力。
(编辑:李 慧)
2016-09-15
谢卫民,男,长江委水文局长江中游水环境监测中心,高级工程师.
1006-0081(2016)11-0062-03
X52
A
