粗粒化模型中的Gay-Berne相互作用势
2016-12-12毛英臣张德鹏刘佳慧王诗佳
毛英臣,张德鹏,刘佳慧,孙 甍,王诗佳
(辽宁师范大学 物理与电子技术学院,辽宁 大连 116029)
粗粒化模型中的Gay-Berne相互作用势
毛英臣,张德鹏,刘佳慧,孙 甍,王诗佳
(辽宁师范大学 物理与电子技术学院,辽宁 大连 116029)
合理描述非球形粗粒化粒子间的相互作用是提高粗粒化分子动力学模拟速度的关键.为此本文介绍了 Gay-Berne势.将之应用于两种有机小分子体系,在合理选择构象集后,由遗传算法得到了粗粒化粒子的 GB参数,并通过对粗粒化模型和全原子模型得到的范德瓦耳斯相互作用的对比检验了GB力场参数.最后,指出如何处理作用位点是粗粒化模型发展的一个关键问题.
粗粒化分子动力学模拟;Gay-Berne势;范德瓦耳斯相互作用;作用位点
对生物大体系和软物质体系的分子模拟而言,随着模拟体系粒子数目的增加,基于微观层次的全原子模拟的运算速度会极速下降,而若采用介观模拟,某些微观特征将会被“模糊”掉,故此介于两种方法间的粗粒化分子模拟常被采用[1].所谓“粗粒化”就是研究体系中的小分子或者小原子集团(如官能团、残基等)视为一个 “粗粒化粒子”,对于“粒子”相互作用则类似于全原子分子力场那样进行构建,运动方程则一般常采用(质点或刚体)牛顿方程.应当指出的是在实际模拟中,对分子间相互作用势的合理描述是准确而高效模拟体系性质的关键所在.本文将介绍目前在粗粒化分子模拟中采用的 Gay-Berne(GB)势[2],并结合两种有机小分子(C5H5N、DMF:C3H7NO)体系进行详细的讨论.
1 Gay-Berne相互作用势
任何一个好的分子模型必须具备两个主要特征,一方面数学上要简单并易于处理,另一方面能够对模拟体系作较好的物理描述[3].基于这两方面的考虑,Berne和 Pechukas近似地将分子视为绕其主轴旋转的椭球体,得到了由高斯函数描述的经验势.在对非球形分子的分子模拟中,短距离吸引和排斥相互作用可通过如下方法来完成,即在分子内定义多个位点,每两个位点间的范德瓦尔斯相互作用势由勒纳德-琼斯势描述.需指出的是对较大分子,计算势的时间会随位点数目的平方而递增,从而使得计算效率大为降低.为了解决这一问题,Gay和 Berne对描述非球形分子间的短距离排斥和吸引相互作用的高斯重叠势进行了修正,把分子处理成软的、单轴的椭球体,把 GB作用位点放置在椭球体的质心上,用单位点势替代多位点势,这样就得到了 GB势[2].在惯性系中,椭球体在位形空间中由其质心坐标和3个欧拉角描述,对应于由一套 GB参数来描述.目前,GB势在描述液晶体系中得到了广泛应用[4-6].为了更好地控制 GB势的软硬度,Brene和Pechukas建议增加参数来控制经验势的柔性,由此引入了 dw参数.
两个粗粒化粒子间的GB势可表示为

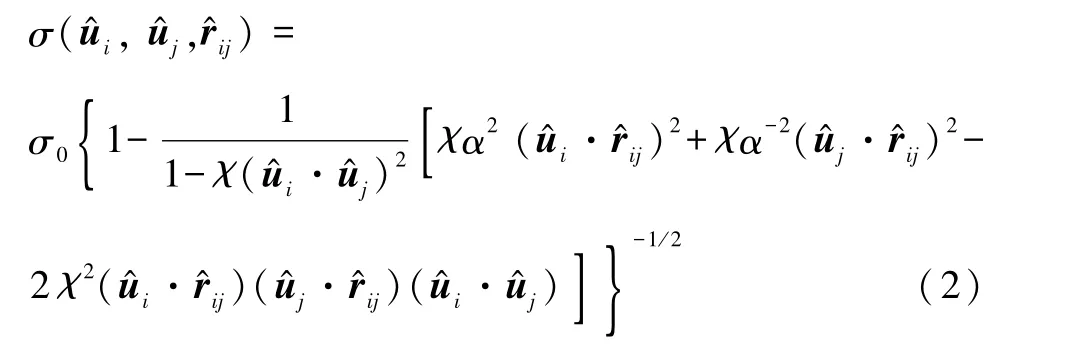
其中
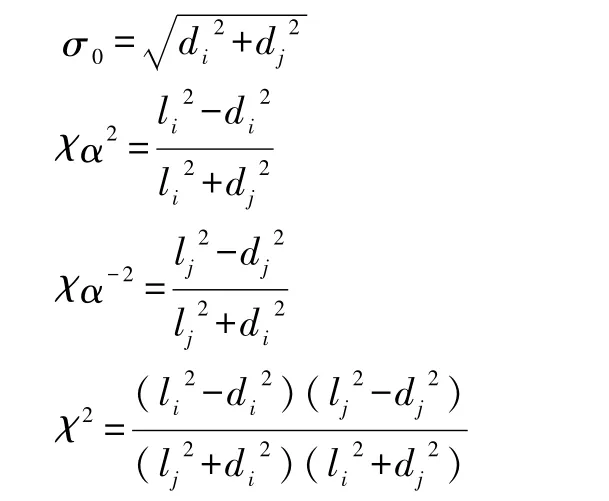
而l和d分别描述分子的长和宽,这样分子的形状就可以用任意的棒状、盘状或者球状等来表示.
式(1)中的阱深参数取如下形式:
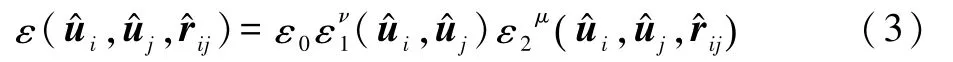
其中μ和ν均为可调指数,借鉴文献[7]工作,本文分别将其取值为2.0和1.0.而ε1和ε2可分别表示为:


式(5)中的χ′和α′分别取如下形式:
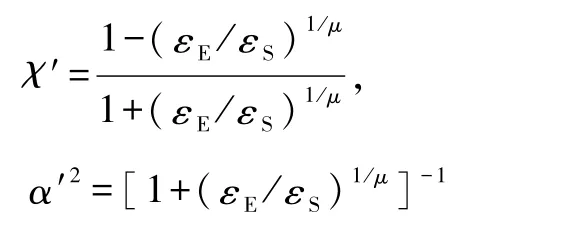
其中 ε0描述了交叉型结构的阱深,εE和 εS分别表示尾对尾和边对边结构的阱深(形象描述参见表1).当两个分子相同或者其中一个分子是球形分子时,势函数用 Berne和 Pechukas的原始形式表示.如果两个分子都是球形的,势函数将进一步简化为勒纳德-琼斯势形式.
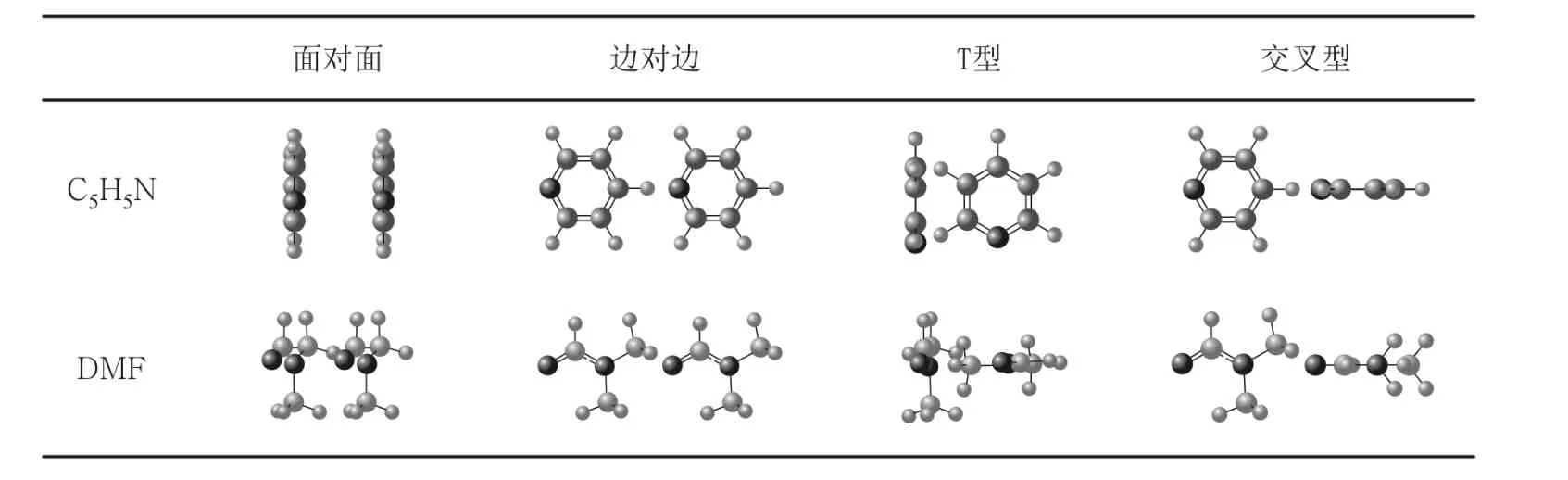
表1 C5H5N和DMF二聚物的4种基本构象
2 粗粒化模型力场参数的确定
为了获得GB势参数,我们需要对研究体系进行合理取样,然后利用描述有机分子的AMBER 力场(GAFF)[8],通过与全原子的范德瓦尔斯相互作用势进行比较后拟合得到GB参数.选取GAFF力场是由于它的函数形式简单,原子类型有限.
在研究中,我们发现构象的选取会直接影响GB参数的拟合,进而决定了GB势是否能够准确地模拟全原子的范德瓦尔斯相互作用势,因此选择可以反映真实分子液体环境的构象是十分重要的.由于在实际的溶液环境中,二聚物的面对面(或尾对尾)、边对边、T型和交叉结构这 4种构象出现的概率相对较大[5],另外考虑到在真实的液体环境中分子的构象趋于取能量最低的构象,所以本文在筛选用于拟合GB参数的构象时,对上述4种构象分别基于玻尔兹曼分布采用了蒙特卡洛方法获得分子的构象集.这样才可以确保在一定的能量范围内,构象数目随能量的升高而减少.表1给出了C5H5N和 DMF二聚物的4种基本构象.
在参数拟合的具体过程中,5个自由参数(l,d,ε0,εE/εS和 dw)由遗传算法拟合得到,两种有机小分子的GB参数见表2.
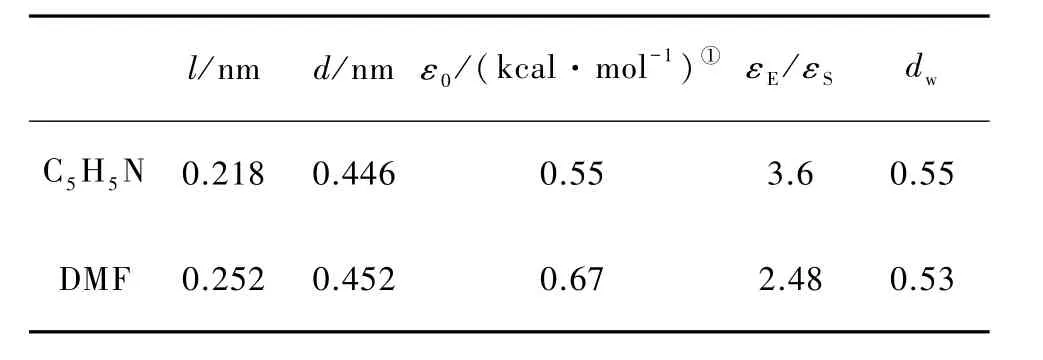
表2 C5H5N和DMF的GB参数
3 在有机小分子体系的应用
为了检验本文对分子粗粒化模型GB参数的拟合,我们对C5H5N、DMF两种有机分子溶剂分别进行了粗粒化的和全原子的分子动力学模拟.为了在计算的过程中可任意地在粗粒化模型和全原子模型间进行转换,方便比较粗粒化模型和全原子模型的结果,故本文在计算中采用了TINKER软件.此外还基于如下考虑:TINKER还包含了对粗粒化粒子的GB相互作用势、力和力矩部分的计算,并可在惯性系中非常容易地整合GB相互作用势的计算结果.两分子间的静电相互作用由电多极展开势(EMP)计算.
C5H5N和 DMF有机溶剂的立方体盒子由Materials Studio(MS)5.0软件生成,其边长均约为30 Å.对GB截断半径都取为12.0 Å(这考虑到在通常情况下,要求截断半径值不大于盒子边长的一半).分子动力学模拟在NVT系综下进行,温度取为298 K.由于自由度的减小,以及分子内高频运动的 “屏蔽”,在粗粒化分子动力学模拟中时间步长取为5 fs.
在图1和图2中,我们比较了利用两种模型计算得到的 C5H5N和 DMF的范德瓦尔斯相互作用势,可以看到粗粒化模型结果整体上较好地符合了全原子模型结果.但对于T型构象,可以看出相比于全原子模型,粗粒化模型得到了较小的阱深和距离参数,同时阱的位置也有一定的偏移.一方面主要是由于粗粒化模型用两个椭球体(长椭球或者扁椭球)并不能很好地表示这两种分子二聚物的T型构象.另一方面,更主要的是我们在计算时,将所用的作用位点都放在其质心(中心)上,而实际上由于原子的电负性(C5H5N中的N原子,DMF中的N原子和O原子)往往会使静电相互作用位点偏离其中心,且由于电多极矩效应,需要用到多个作用位点.对比图1与图2,我们还发现DMF的这种偏移要较C5H5N更大.产生这样结果的原因是,对于DMF分子而言,静电作用位点与GB作用位点的距离要相较C5H5N分子更大,所以造成了势阱位置的更大偏移.由图1和图2我们还可看到,对于二聚物的边靠边型和交叉型两种构象,利用两种模型得到的范德瓦尔斯相互作用非常接近,这同样是由于在目前的计算中仅考虑了单作用位点的原因.对于上述问题,文献[9]提出了多作用位点的GB-EMP模型,很好地改进了计算结果.

图1 基于粗粒化模型和全原子模型对 C5H5N分子的范德瓦尔斯相互作用曲线的比较.二聚物的4种构象:面对面(1、2),边对边(7、8),交叉型(5、6)和 T形(3、4).实线代表了粗粒化模型结果,虚线代表了全原子模型结果.
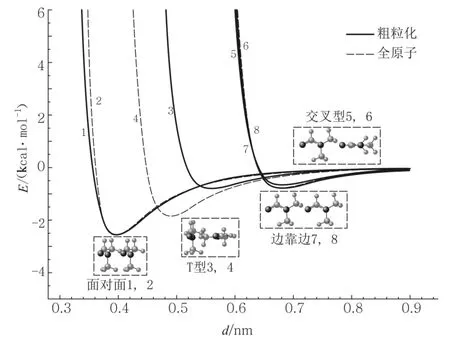
图2 基于粗粒化模型和全原子模型对 DMF分子范德瓦尔斯相互作用曲线的比较(同图1)
4 结论
加快分子动力学模拟速度的一种有效的办法是采用粗粒化模型,为此需要合理且高效地描写粗粒化粒子间的相互作用.一般地粗粒化粒子都不是球形的,为了精确描述非球形分子间的范德瓦耳斯相互作用,Gay-Berne势将原来的球形分子改进为对称椭球形分子.本文详细地介绍了Gay-Berne势,首先对C5H5N和DMF两种分子的二聚体进行了合理取样,然后由遗传算法拟合得到了两种分子的粗粒化粒子的GB参数.为了检验所获得的GB参数,我们对两种有机小分子溶剂进行了分子动力学模拟,并同全原子模拟结果进行了比较.粗粒化模型结果较好地符合了全原子模型结果.但是对于T型构象,由于本工作将所有作用位点放在粗粒化粒子的中心上,所以势阱参数和位置有了较大的偏差.更准确地描述粒子间的相互作用,需考虑多作用位点,但应当指出的是作用位点的增加意味着计算速度的降低,因此如何平衡计算速度和精度是粗粒化模型发展必须考虑的一个问题.
[1] Voth G A.Coarse-Graining of Condensed Phase and Biomolecular Systems[M].Boca Raton:CRC Press,2009.
[2] Gay J G,Berne B J.Modification of the overlap potential to mimic a linear site-site potential[J].Journal of Chemical Physics,1981,74:3316-3319.
[3] Berne B J,Pechukas P.Gaussian model potentials for molecular interactions[J].Journal of Chemical Physics,1972,56:4213-4216.
[4] Cleaver D J,Care C M,Allen M P,et al.Extension and generalization of the Gay-Berne potential[J].Physical Review E,1996,54:559-567.
[5] Care C M,Cleaver D J.Computer simulation of liquid crystals[J].Reports on Progress in Physics,2005,68:2665-2700.
[6] 陈正隆,徐为人,汤立达.分子模拟的理论与实践 [M].北京:化学工业出版社,2007:18-23.
[7] Golubkov P A,Ren P Y.Generalized coarse-grained model based on point multipole and Gay-Berne potentials[J].Journal of Chemical Physics,2006,125:064103.
[8] Wang J M,Wolf R M,Caldwell W J,et al.Development and testing of a general AMBER force field[J].Journal of Computational Chemistry,2004,25:1157-1174.
[9] Golubkov P A,Wu J C,Ren P Y.A transferable coarsegrained model for hydrogen-bonding liquids[J].Physical Chemistry Chemical Physics,2008,10:2050-2057.
Gay-Berne interaction potential used in coarse-graining molecular simulation
MAO Ying-chen,ZHANG De-peng,LIU Jia-hui,SUN Meng,WANG Shi-jia
(School of Physics and Electronic Technology,Liaoning Normal University,Dalian,Liaoning 116029,China)
How to compute effectively the interactions of non-spherical coarse-graining particles is critical for the improvement of computational speed of the coarse-grained molecular simulation,so we introduce Gay-Berne potential in this paper,and carry out applications for two small organic molecular systems.We derive the GB parameters of coarse-graining particles with genetic algorithm based on the reasonable conformational set.Moreover,we make some tests about the GB parameters through making comparison with the results from coarse-grained model and all-atom model,respectively.Finally,we point out how to deal with the interaction site is an essential problem for the development of coarse-graining model.
coarse-grained molecular dynamics simulation;Gay-Berne interaction;van der Waals potential;interaction site
教学讨论
O 469
A
1000-0712(2016)10-0020-03
2015-12-05;
2016-02-25
国家自然科学基金项目(11447019)、教育部高等学校力学课程教学研究项目(JZW-15-LX-18)资助
毛英臣(1977—),男,山东莱州人,博士,辽宁师范大学物理与电子技术学院副教授,主要从事理论物理的研究和教学工作.
