Time- and cell-type specific changes in iron, ferritin, and transferrin in the gerbil hippocampal CA1 region after transient forebrain ischemia
2016-12-02DaeYoungYooKiYeonYooJoonHaParkHyunJungKwonHyoYoungJungJongWhiKimGoangMinChoiSeungMyungMoonDaeWonKimYeoSungYoonMooHoWonInKooHwang
Dae Young Yoo, Ki-Yeon Yoo, Joon Ha Park, Hyun Jung Kwon, Hyo Young Jung, Jong Whi Kim, Goang-Min Choi, Seung Myung Moon, Dae Won Kim, Yeo Sung Yoon, Moo-Ho Won, In Koo Hwang,
1 Department of Anatomy and Cell Biology, College of Veterinary Medicine, and Research Institute for Veterinary Science, Seoul National University, Seoul, South Korea
2 Department of Oral Anatomy, Research Institute of Oral Sciences, College of Dentistry, Gangneung-Wonju National University, Gangneung, South Korea
3 Department of Neurobiology, School of Medicine, Kangwon National University, Chuncheon, South Korea
4 Department of Biochemistry and Molecular Biology, Research Institute of Oral Sciences, College of Dentistry, Gangneung-Wonju National University, Gangneung, South Korea
5 Department of Thoracic and Cardiovascular Surgery, Chuncheon Sacred Heart Hospital, College of Medicine, Hallym University, Chuncheon, South Korea
6 Department of Neurosurgery, Dongtan Sacred Heart Hospital, College of Medicine, Hallym University, Hwaseong, South Korea
RESEARCH ARTICLE
Time- and cell-type specific changes in iron, ferritin, and transferrin in the gerbil hippocampal CA1 region after transient forebrain ischemia
Dae Young Yoo1, Ki-Yeon Yoo2, Joon Ha Park3, Hyun Jung Kwon4, Hyo Young Jung1, Jong Whi Kim1, Goang-Min Choi5, Seung Myung Moon6, Dae Won Kim4, Yeo Sung Yoon1, Moo-Ho Won3, In Koo Hwang1,*
1 Department of Anatomy and Cell Biology, College of Veterinary Medicine, and Research Institute for Veterinary Science, Seoul National University, Seoul, South Korea
2 Department of Oral Anatomy, Research Institute of Oral Sciences, College of Dentistry, Gangneung-Wonju National University, Gangneung, South Korea
3 Department of Neurobiology, School of Medicine, Kangwon National University, Chuncheon, South Korea
4 Department of Biochemistry and Molecular Biology, Research Institute of Oral Sciences, College of Dentistry, Gangneung-Wonju National University, Gangneung, South Korea
5 Department of Thoracic and Cardiovascular Surgery, Chuncheon Sacred Heart Hospital, College of Medicine, Hallym University, Chuncheon, South Korea
6 Department of Neurosurgery, Dongtan Sacred Heart Hospital, College of Medicine, Hallym University, Hwaseong, South Korea
In the present study, we used immunohistochemistry and western blot analysis to examine changes in the levels and cellular localization of iron, heavy chain ferritin (ferritin-H), and transferrin in the gerbil hippocampal CA1 region from 30 minutes to 7 days following transient forebrain ischemia. Relative to sham controls, iron reactivity increased significantly in the stratum pyramidale and stratum oriens at 12 hours following ischemic insult, transiently decreased at 1—2 days and then increased once again within the CA1 region at 4—7 days after ischemia. One day after ischemia, ferritin-H immunoreactivity increased significantly in the stratum pyramidale and decreased at 2 days. At 4—7 days after ischemia, ferritin-H immunoreactivity in the glial components in the CA1 region was significantly increased. Transferrin immunoreactivity was increased significantly in the stratum pyramidale at 12 hours, peaked at 1 day, and then decreased significantly at 2 days after ischemia. Seven days after ischemia, Transferrin immunoreactivity in the glial cells of the stratum oriens and radiatum was significantly increased. Western blot analyses supported these results, demonstrating that compared to sham controls, ferritin H and transferrin protein levels in hippocampal homogenates significantly increased at 1 day after ischemia, peaked at 4 days and then decreased. These results suggest that iron overload-induced oxidative stress is most prominent at 12 hours after ischemia in the stratum pyramidale, suggesting that this time window may be the optimal period for therapeutic intervention to protect neurons from ischemia-induced death.
nerve regeneration; ischemia; iron; ferritin heavy chain; transferrin; CA1 region; oxidative stress; neural regeneration
orcid: 0000-0002-0533-4638 (In koo Hwang)
Accepted: 2016-04-28
Introduction
Transient forebrain ischemia, an acute interruption or occlusion of cerebral vessels, ranks among the leading causes of death and severe disability. The affected regions experience various pathophysiological events, such as changes in ion homeostasis, inflammation, and oxidative stress, in the post-ischemic stages (Lipton, 1999). In particular, reactive oxygen species (ROS) and their intermediates, which are induced by reperfusion, adversely affect many cellular functions.
Iron plays important roles in the formation of myelin, synthesis and metabolism of neurotransmitters, and development of neuronal dendritic tree under normal physiological circumstances and at various developmental stages (Ben-Shachar et al., 1986; Youdim et al., 1991; Crichton, 2009). Deficiency of iron during early development affects hippocampal function in both animal models and humans (Lozoff and Georgieff, 2006; Geng et al., 2015). Conversely, excess iron levels generate ROS via the Harber-Weiss reaction, which can damage cellular membranes, proteins, and DNA (Halliwell and Gutteridge, 1984; Sorond and Ratan, 2000). Neurons are especially susceptible to ROS damage due totheir high content of polyunsaturated fatty acids and iron but relatively low levels of antioxidants (Halliwell, 2006).
Sources of iron include extracellular fluid, which is potentially involved in both intracellular iron storage and intravascular iron transport and delivery to the brain (Lipscomb et al., 1998; Ding et al., 2011). Transferrin contains two specific high-affinity Fe3+binding sites and moves the peripheral cellular iron into cells by binding to transferrin receptor 1. In particular, ferritin heavy chain (ferritin-H) maintains cellular iron balance via the regulation of reuptake and storage of Fe3+(Harrison and Arosio, 1996; Chasteen, 1998). In addition, ferritin-H may be a reactant protein to oxidative stress (Lin and Girotti, 1997; Garner et al., 1998; Oberle et al., 1998). Several lines of evidence demonstrate that iron concentration, and consequent alterations of ferritin and transferrin, increase with age and some neurological disorders (Ishimaru et al., 1996; Bishop and Robinson, 2001; Youdim et al., 2004; Zecca et al., 2004; Moos et al., 2007; Yoo et al., 2007; Snyder and Connor, 2009; Li et al., 2012).
Previous studies have generally used biochemical approaches to measure changes in iron, ferritin, and transferrin in the brain following ischemic insult. However, examining these molecules at the cellular level may provide further clues on how to protect neurons from ischemic damage. In the present study, we investigated cell-specific changes in iron, ferritin-H, and transferrin levels in the hippocampal CA1 region of gerbils at multiple time points following transient ischemia.
Materials and Methods
Experimental animals
Male gerbils (n = 70, 3-month-old, 50—60 g) were purchased from Japan SLC Inc. (Shizuoka, Japan). They were housed under standard conditions with adequate temperature (22°C) and humidity (60%) control, a 12-hour light/dark cycle, and free access to food and water, as previously described (Yoo et al., 2012a). The handling and care of the animals conformed to guidelines compliant with current international laws and policies (NIH Guide for the Care and Use of Laboratory Animals, NIH Publication No. 85-23, 1985, revised 1996). Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University, South Korea. All experiments were conducted with an effort to minimize the number of animals used and the suffering caused by the procedures employed in the present study.
Induction of transient forebrain ischemia
As previously described (Yoo et al., 2012a), the animals were anesthetized with a mixture of 2.5% isoflurane (Baxtor, Deerfield, IL, USA) in 33% oxygen and 67% nitrous oxide. Common carotid arteries from both sides were isolated and occluded using non-traumatic aneurysm clips. The complete interruption of blood flow was confirmed by observing the central artery in retinas using an ophthalmoscope (HEINE K180®, Heine Optotechnik, Herrsching, Germany). After 5 minutes of occlusion, the aneurysm clips were removed. Body temperature under free-regulating or normothermic (37 ± 0.5°C) conditions was monitored with a rectal temperature probe (TR-100; Fine Science Tools, Foster City, CA, USA) and maintained using a thermometric blanket before, during, and after the surgery until the animals completely recovered from anesthesia. Thereafter, animals were kept in a thermal incubator (Mirae Medical Industry, Seoul, South Korea) to maintain body temperature until the animals were euthanized. Six animals were excluded due to incomplete occlusion of the common carotid arteries and uncontrolled body temperature.
Tissue processing
As previously described (Yoo et al., 2012b), the animals in the sham-operated (n =5) and ischemia groups (n = 40) were anesthetized with 1 g/kg urethane (Sigma-Aldrich, St. Louis, MO, USA) at various time points (30 minutes, 3, 6, 12 hours, 1, 2, 4, 7 days, n = 5 per group) after surgery. Animals were perfused transcardially with 0.1 M phosphate-buffered saline (PBS, pH 7.4), followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4). Brains were removed and postfixed in the same fixative for 12 hours before undergoing cryoprotection via overnight storage in 30% sucrose. Serial coronal brain sections (30 µm) were generated using a cryostat (Leica, Wetzlar, Germany) and collected into 6-well plates containing PBS. To ensure that the histochemical and immunohistochemical data were comparable between groups, sections were carefully processed under parallel conditions. Tissue sections located 90 µm apart from each other were selected from an area between 1.4 mm and 2.0 mm posterior to the bregma, as defined by a gerbil atlas (Loskota et al., 1974).
Histochemistry for iron
According to a previously described method (Hill and Switzer, 1984; Yoo et al., 2007), five sections from each group were incubated in Perl's solution (1:1, 2% HCl and 2% potassium ferrocyanide) at room temperature for 30 minutes, rinsed in deionized water for 30 minutes, and then developed for at least 15 minutes in 0.5% 3,3′-diaminobenzidine tetrachloride (DAB; Sigma-Aldrich, St. Louis, MO, USA) in PBS (pH 7.4). Sections were rinsed with deionized water for 30 minutes, dehydrated through a graded ethanol series and mounted with Canada balsam (Kanto, Tokyo, Japan).
Immunohistochemistry
As previously described (Yoo et al., 2012b), five tissue sections located 90 µm apart from each other were sequentially incubated with 0.3% hydrogen peroxide (H2O2) in PBS for 30 minutes and 10% normal goat serum in 0.05 M PBS for 30 minutes. Sections were then incubated with a rabbit anti-ferritin-H antibody (diluted 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or rabbit anti-transferrin antibody (diluted 1:200; Santa Cruz Biotechnology) overnight at room temperature. Sections were then incubated with biotinylated goat anti-rabbit IgG for 2 hours, followed by a streptavidin-peroxidase complex for 1 hour at room temperature (1:200; Vector, Burlingame, CA, USA). Immunostaining wasvisualized by reaction with DAB in 0.1 M Tris-HCl buffer (pH 7.2). Sections were dehydrated and mounted on gelatin-coated slides in Canada balsam (Kanto).
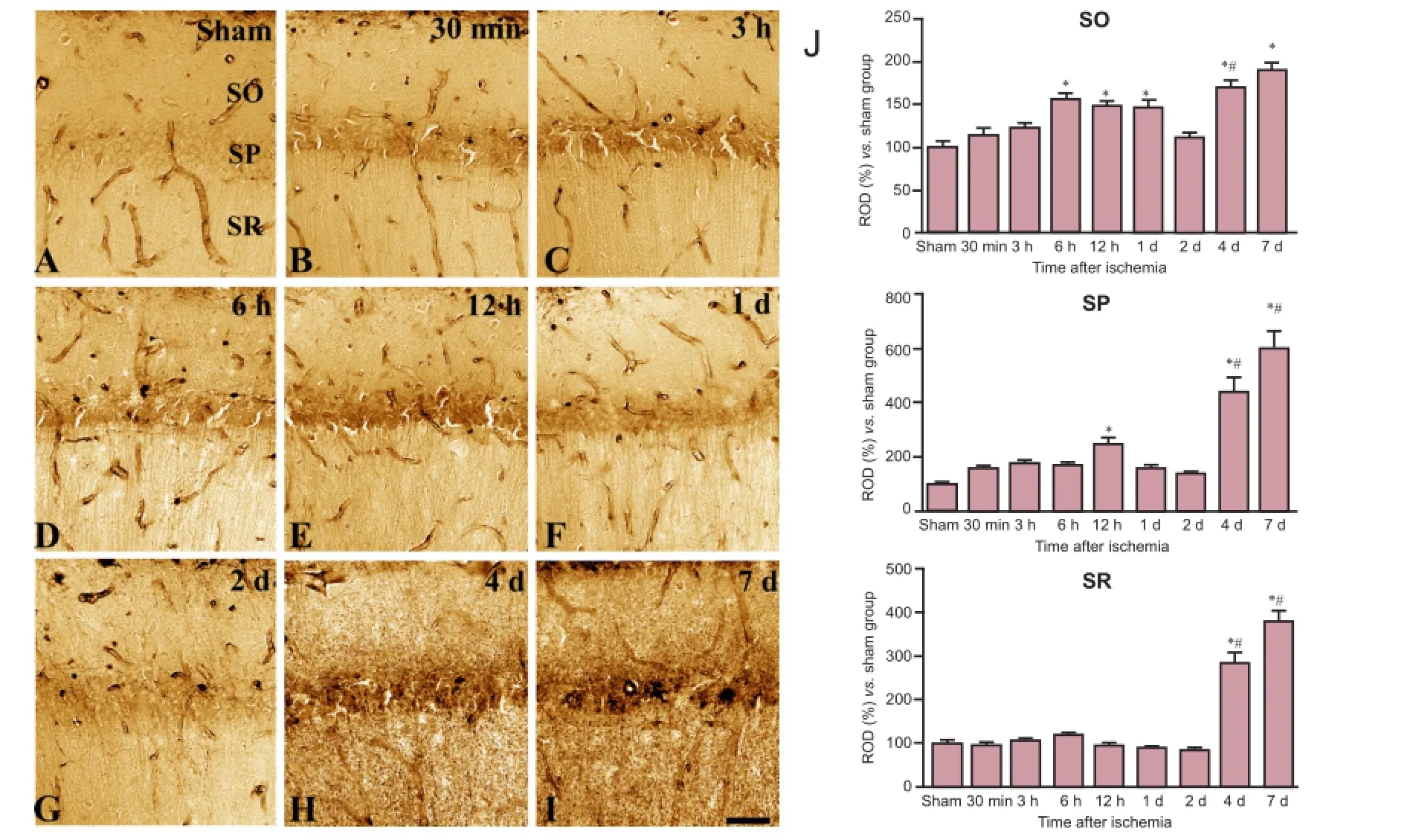
Figure 1 Histochemical staining for iron in the hippocampal CA1 region in sham-operated (sham; A) and ischemia (B—I) groups.
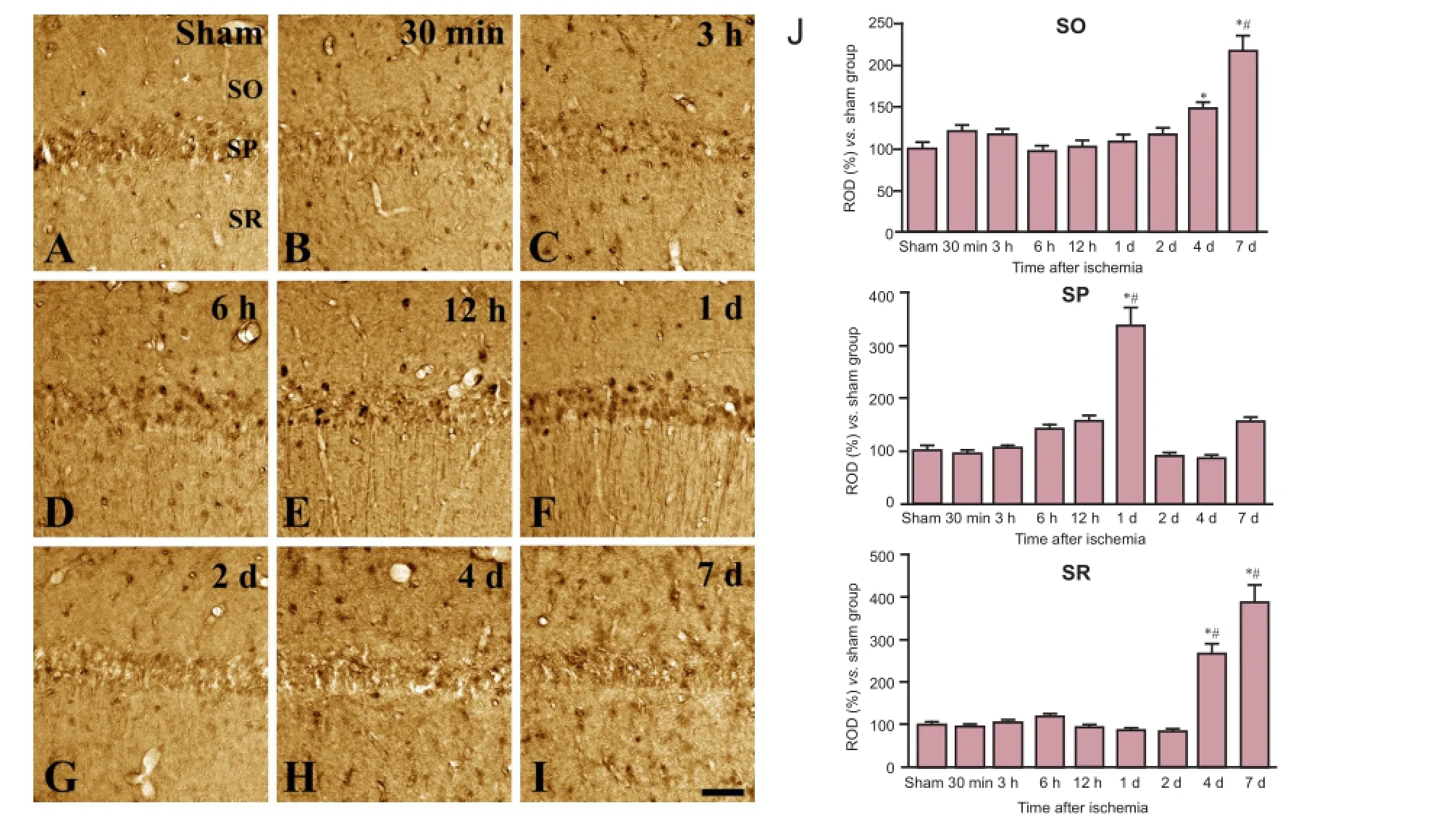
Figure 2 Immunohistochemical staining for ferritin-H in the hippocampal CA1 region in sham-operated (sham; A) and ischemia (B—I) groups.
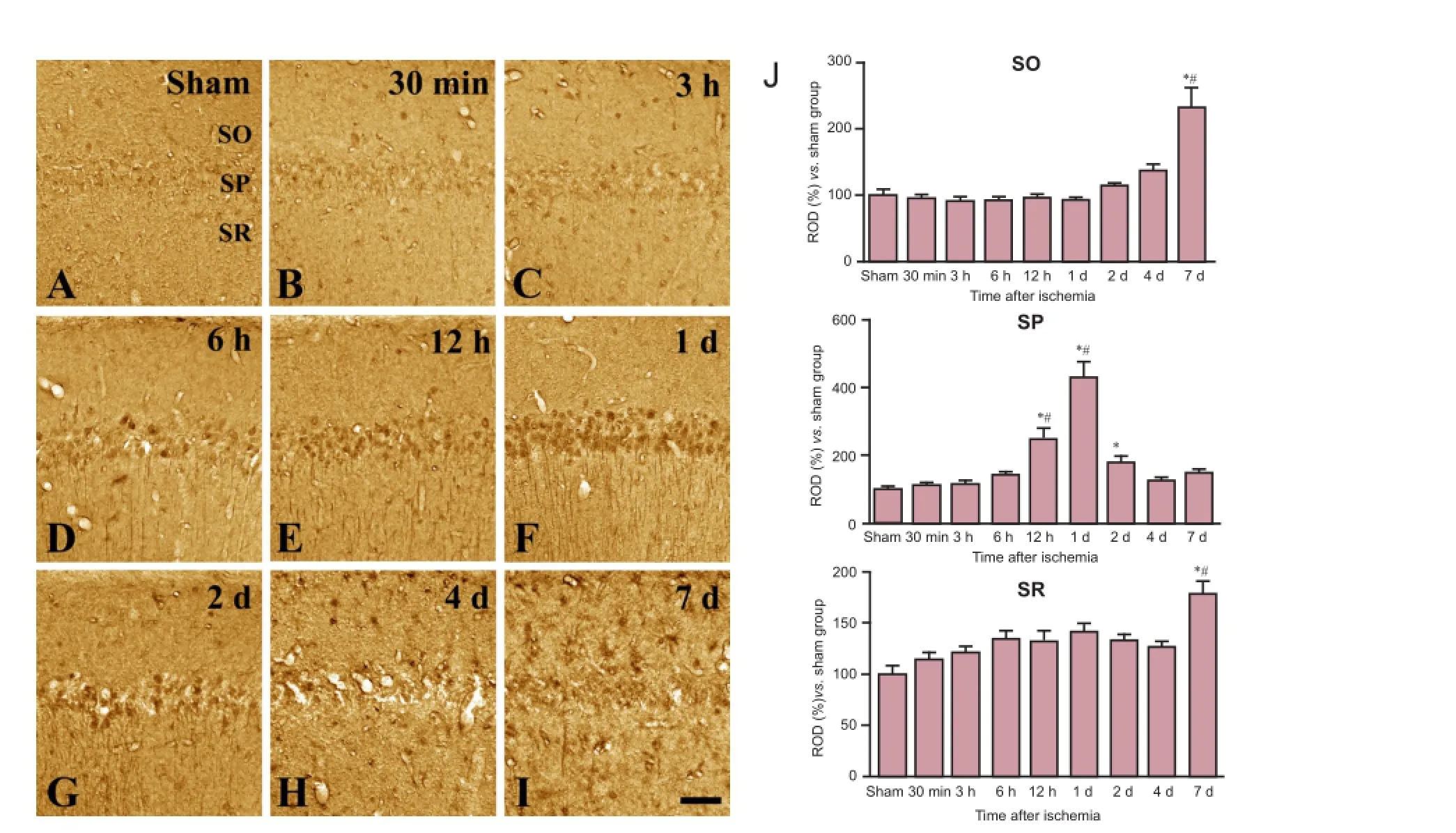
Figure 3 Immunohistochemical staining for transferrin in the CA1 region in sham-operated (sham; A) and ischemia (B—I) groups.
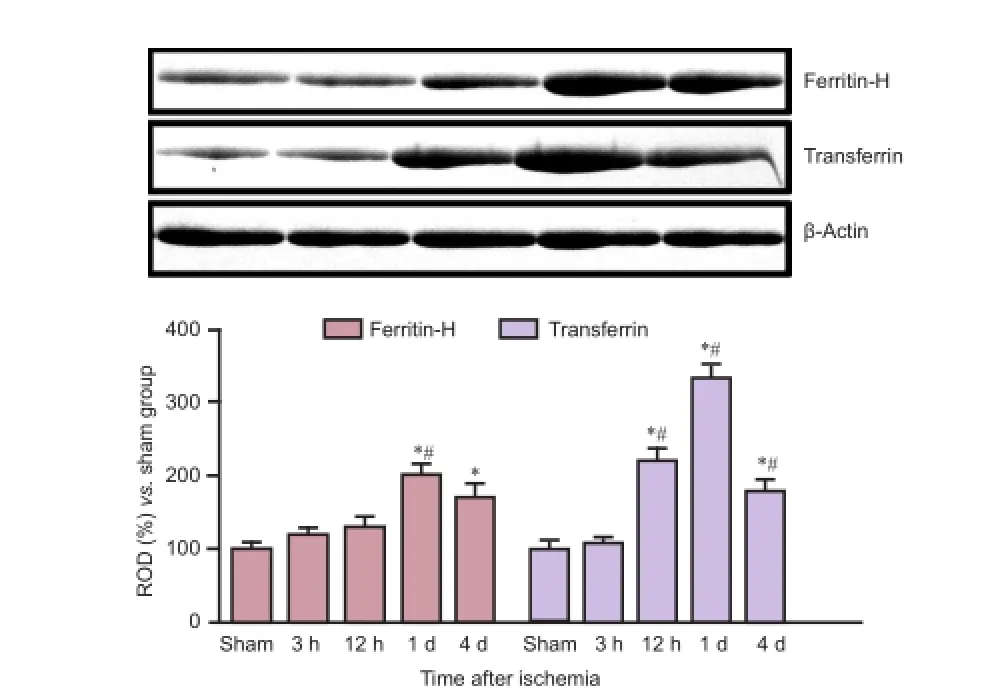
Figure 4 Ferritin-H and transferrin levels in the hippocampus after ischemia.
Data analysis
Analysis of the hippocampal CA1 region was performed using an image analysis system and ImageJ software v. 1.5 (National Institutes of Health, Bethesda, MD, USA), according to a previously described method (Jung et al., 2016). Digital images of the mid-point of the hippocampal CA1 region were captured with a BX51 light microscope (Olympus, Tokyo, Japan) equipped with a digital camera (DP72, Olympus) connected to a computer monitor. Images were calibrated into an array of 512 × 512 pixels corresponding to a tissue area of 1,200 µm × 900 µm (100× primary magnification). Each pixel resolution was 256 gray levels, and the area was divided into strata oriens, pyramidale, and radiatum. The intensity of iron reactivity and ferritin-H and transferrin immunoreactivity were evaluated by relative optical density (ROD), which was obtained after transformation of the mean gray level using the formula: ROD = log(256/mean gray level). ROD of background staining was determined in unlabeled portions of the sections using Photoshop CC software (Adobe Systems Inc., San Jose, CA, USA), and this value was subtracted to correct for nonspecific staining, using ImageJ v. 1.50 software (National Institutes of Health). Data are expressed as a percentage of the sham-operated group values (set to 100%).
Western blot analysis
To quantify changes in ferritin-H and transferrin levelswithin the hippocampus, animals were euthanized at 3, 12 hours, 1 and 4 days after ischemia or following the sham operation (n = 5 animals per group) and their brains removed. Tissues were dissected for use in western blot analysis, conducted as previously described (Yoo et al., 2012a). In brief, 500 µm thick sections of each brain were generated using a vibratome (Leica Microsystems, GmbH, Germany), and the hippocampal area was subdissected using a surgical blade. Hippocampal tissues were homogenized in 50 mM PBS (pH 7.4), containing 0.1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (pH 8.0); 0.2% Nonidet P-40; 10 mM ethylenediaminetetraacetic acid (pH 8.0); 15 mM sodium pyrophosphate; 100 mM β-glycerophosphate; 50 mM NaF; 150 mM NaCl; 2 mM sodium orthovanadate; 1 mM phenylmethylsulfonyl fluoride; and 1 mM dithiothreitol (DTT). After centrifugation, protein levels in the supernatants were determined using a Micro BCA protein assay kit according to the manufacturer's instructions (Pierce Chemical, Rockford, IL, USA). Aliquots containing 20 µg of total protein were denatured by boiling in loading buffer containing 150 mM Tris (pH 6.8), 3 mM DTT, 6% sodium dodecyl sulfate, 0.3% bromophenol blue, and 30% glycerol. Each aliquot was loaded onto a polyacrylamide gel. After electrophoresis, proteins were transferred to nitrocellulose membranes (Pall Crop, East Hills, NY, USA), which were then blocked in 5% non-fat dry milk in PBS/0.1% Tween 20 for 45 minutes, prior to incubation with a peroxidase-conjugated rabbit anti-ferritin-H antibody (diluted 1:500) or a rabbit anti-transferrin antibody (diluted 1:1,000) overnight. Detection was performed for 2 hours after addition of peroxidase-conjugated anti-rabbit IgG using an enhanced luminol-based chemiluminescent kit (Pierce Chemical). The blots were scanned, and densitometry was performed for the quantification of relative density of each band, using Scion Image software (Scion Corp., Frederick, MD, USA). Blots were stripped and reprobed with an antibody against β-actin as an internal loading control. Data were normalized to β-actin level in each lane.
Statistical analysis
As previously described (Yoo et al., 2012b; Jung et al., 2016), all data are shown as the mean ± SEM. Differences among the means were statistically analyzed by one-way analysis of variance followed by a Bonferroni post hoc test, using Graph-Pad Prism 5.01 software (GraphPad Software, Inc., La Jolla, CA). Statistical significance was considered at P < 0.05.
Results
Changes in iron reactivity in the hippocampal CA1 region In the sham-operated group, iron reactivity was mainly detected in blood vessels, with faint iron reactivity also observed in the stratum pyramidale of the CA1 region (Figure 1A). In contrast, in the animals which received a transient ischemic insult, iron reactivity increased in the CA1 region from 30 minutes to 12 hours after ischemia, peaked in the stratum pyramidale at 12 hours after ischemia (Figure 1B—D, J). At later time points after ischemic insult, iron reactivity exhibited a biphasic pattern, with levels significantly decreased at 1 and 2 days compared to 12 hours after ischemia (Figure 1E, F, J) but elevated again at 4 and 7 days after ischemia (Figure 1G, H, J). At 4 and 7 days after ischemia, iron reactivity was observed in all CA1 regions, including the strata oriens, pyramidale, and radiatum.
Changes in ferritin-H immunoreactivity in the hippocampal CA1 region
In the sham-operated group, a few ferritin-H immunoreactive cells were detected in the CA1 region of the hippocampus (Figure 2A). In the experimental group, ferritin-H immunoreactivity was detected at a level similar to controls at 30 minutes and 3 hours after ischemia (Figure 2B, C, J). Ferritin-H immunoreactivity was significantly increased at 6 hours after ischemia and remained elevated till 1 day after ischemia. Highest ferritin-H immunoreactivity in the stratum pyramidale appeared at 6, 12 and 1 day after ischemia (Figure 2D, E, and J). By 2 days after ischemia, ferritin-H immunoreactivity decreased significantly relative to peak levels and was no longer detectable in the stratum pyramidale (Figure 2G, J). Four days after ischemia, ferritin-H immunoreactivity was observed in glial cells located in strata oriens and radiatum (Figure 2H, J) and was further increased in these layers by 7 days (Figure 2I, J). Western blot analysis showed that ferritin H protein levels in ischemic brain were similar to sham-control levels at 3 and 12 hours but were significantly increased at 1 day after ischemia. Ferritin-H protein level decreased relative to peak values at 4 days after ischemia, but it was still significantly higher than that in the sham-operated group (Figure 4).
Changes in transferrin immunoreactivity in the hippocampal CA1 region
In the sham-operated group, weak transferrin immunoreactivity was detected in a few cells located in the strata oriens and radiatum (Figure 3A). In the experimental group, the immunoreactivity and distribution pattern of transferrin at 30 minutes to 3 hours after ischemia were similar to those in the sham-operated group (Figure 3B, C, J). Transferrin immunoreactivity started to increase in the stratum pyramidale of the CA1 region at 6 hours after ischemia (Figure 3D, E, J) and was most pronounced in the stratum pyramidale at 1 day after ischemia (Figure 3F, J). By 2 days after ischemia, transferrin immunoreactivity decreased in the CA1 region relative to that at 1 day but it remained at significantly higher level than that in the sham-operated group (Figure 3G, J). By 4 days after ischemia, a few transferrin-immunoreactive glial cells were detected in the strata oriens and radiatum (Figure 3H, J), and transferrin immunoreactivity in the CA1 region significantly increased over this level by 7 days after ischemia (Figure 3I, J). Similar to these results, western blot analysis showed that transferrin protein level was unchanged in hippocampal samples compared to controls at 3 hours after ischemia but significantly increased at 1 day after ischemia. Transferrin protein level decreased from peak level at 4 days after ischemia, but it remained higher than that in thesham-operated group (Figure 4).
Discussion
Iron is the most abundant trace metal in the brain. Iron overload during hypoxia and reperfusion appears to accelerate the generation of ROS, such as superoxide anion and hydrogen peroxide, by the iron-dependent conversion into reactive hydroxyl radicals, which causes damage to membrane, protein, and DNA, and subsequent neuronal death (Halliwell and Gutteridge, 1984; Sorond and Ratan, 2000; Connor and Ghio, 2009; Adibhatla and Hatcher, 2010). However, the exact mechanism involved and the cell-specific changes that occur in iron and its regulatory proteins following ischemia are not understood. In the present study, we examined the spatiotemporal expression patterns of iron and its related proteins in response to ischemic insult. We measured iron reactivity by Perl's method, which showed biphasic increases in iron levels in the hippocampal CA1 region in response to ischemia. This result is consistent with previous studies also using Perl's method that showed strong reactivity in pyramidal cell bodies and dendrites of the CA1 region and in cortical cells of the cerebral hemispheres in experimental animals subjected to insult in chronic hypoperfusion and four-vessel occlusion models of ischemia (Kondo et al., 1995; Li et al., 2012). However, in the present study, we observed transient decreases in iron reactivity 1—2 days after ischemia. This reduction in iron reactivity may be associated with a compensatory increase in antioxidants such as copper-zinc superoxide dismutase (SOD1). In a previous study, we observed that SOD1 immunoreactivity in the stratum pyramidale increased significantly at 1 day after ischemia and then decreased (Hwang et al., 2005). This transient increase in SOD1 following ischemic insult may decrease iron-catalyzed oxidative damage, which results from iron ions forming highly reactive hydroxyl radicals, which are thought to facilitate lipid peroxidation of the cell membrane and, ultimately, cause cell death (Halliwell, 1992; Bishop and Robinson, 2001). In support of this hypothesis, the administration of deferoxamine, an iron chelator capable of permeating the blood-brain barrier, protects neurons from ischemic injury (Siddig et al., 2009; Yang et al., 2011).
Next, we observed that ferritin-H immunoreactivity peaked in the CA1 region at 1 day after ischemia. This result is partially supported by previous studies that found increased levels of free iron and ferritin in ischemic brain (Lipscomb et al., 1998; Chi et al., 2000; Ding et al., 2011). However, Chi et al. (2000) demonstrated that the elevation of ferritin-H mRNA was maintained for up to 336 hours after the onset of reperfusion. In the present study, we observed increases of ferritin-H in the stratum pyramidale at 1 day after ischemia and in the glial components at 4—7 days after ischemia. The early increase of ferritin-H may serve to sequester excess iron to protect pyramidal cells against iron toxicity (Arosio and Levi, 2010), and the latter increase may be associated with glial sequestration of iron. This result is consistent with the observed transient peak in iron reactivity at 12 hours following ischemia. In rat four-vessel occlusion model of ischemia, iron begins to accumulate at 4 weeks after recirculation, and ferritin-immunoreactive microglia have been found between 4—24 weeks after recirculation (Kondo et al., 1995). In the current study, we also observed significantly increased transferrin immunoreactivity in the stratum pyramidale at 1 day after ischemia and in the glial components in the strata oriens and radiatum at 4—7 days after ischemia.
In the present study, we observed a significant increase of transferrin which occurred in a biphasic pattern in the hippocampal CA1 region, e.g., in the stratum pyramidale before neuronal death is known to occur (1 day after ischemia) and in strata oriens and radiatum 7 days after ischemia. Transferrin receptor levels were also found to be significantly elevated in the hippocampus and cortex after chronic cerebral hypoperfusion (Li et al., 2012) and middle cerebral artery occlusion (Ding et al., 2011). In a previous study using the gerbil ischemia model, transferrin immunoreactivity in the hippocampal CA1 region of experimental animals at 1 day post-ischemia was similar to that of sham controls and was significantly increased at 7 days after ischemia (Ishimaru et al., 1996). However, this previous study did not observe changes in ferritin and transferrin immunoreactivity in the hippocampal CA1 region. This discrepancy may be associated with the anesthetics used and rectal temperature differences between the two studies and, consequently, vulnerability to ischemic damage. We set the rectal temperature as 37°C and used isoflurane as an anesthetic, whereas Ishmaru et al. (1996) used halothane at 38°C. Kil et al. (1996) reported that hyperthermia significantly increased salicylate hydroxylation products in the brain compared to that under normothermic conditions. In addition, isoflurane-anesthetized animals show less degradation of microtubule-associated protein 2 in the hippocampus than halothane-anesthetized animals (Sugaya and Kitani, 1993).
In conclusion, our findings suggest that the transient increases in ferritin and transferrin may act to compensate for iron overload. However, the increases of ferritin and transferrin are not sufficient to protect neurons from ischemic damage. Nevertheless, these transient increases in protein suggest that there might be a window of opportunity for therapeutic interventions to protect neurons from oxidative damage induced by ischemia.
Author contributions: DYY and IKH wrote the paper. DYY, KYY, JHP, HYJ and JWK conducted the histochemistry and immunohistochemistry, and analyzed the data. DYY and KYY prepared animal models. GMC, SMM and YSY designed the experiments. MHW performed the experiments and revised the paper. HJK and DWK conducted western blot analysis for ischemic tissues. All authors approved the final version of the paper.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
References
Adibhatla RM, Hatcher JF (2010) Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 12:125-169.
Arosio P, Levi S (2010) Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 1800:783-792.
Ben-Shachar D, Ashkenazi R, Youdim MB (1986) Long-term consequence of early iron-deficiency on dopaminergic neurotransmission in rats. Int J Dev Neurosci 4:81-88.
Bishop GM, Robinson SR (2001) Quantitative analysis of cell death and ferritin expression in response to cortical iron: implications for hypoxia-ischemia and stroke. Brain Res 907:175-187.
Chasteen ND (1998) Ferritin. Uptake, storage, and release of iron. Met Ions Biol Syst 35:479-514.
Chi SI, Wang CK, Chen JJ, Chau LY, Lin TN (2000) Differential regulation of H- and L-ferritin messenger RNA subunits, ferritin protein and iron following focal cerebral ischemia-reperfusion. Neuroscience 100:475-484.
Connor JR, Ghio AJ (2009) The impact of host iron homeostasis on disease. Preface. Biochim Biophys Acta 1790:581-582.
Crichton RR (2009) Inorganic biochemistry of iron metabolism from molecular mechanisms to clinical consequences. Chichester: John Wiley & Sons.
Ding H, Yan CZ, Shi H, Zhao YS, Chang SY, Yu P, Wu WS, Zhao CY, Chang YZ, Duan XL (2011) Hepcidin is involved in iron regulation in the ischemic brain. PLoS One 6:e25324.
Garner B, Roberg K, Brunk UT (1998) Endogenous ferritin protects cells with iron-laden lysosomes against oxidative stress. Free Radic Res 29:103-114.
Geng F, Mai X, Zhan J, Xu L, Zhao Z, Georgieff M, Shao J, Lozoff B (2015) Impact of fetal-neonatal iron deficiency on recognition memory at 2 months of age. J Pediatr 167:1226-1232.
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634-1658.
Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59:1609-1623.
Halliwell B, Gutteridge JM (1984) Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J 219:1-14.
Harrison PM, Arosio P (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161-203.
Hill JM, Switzer RC 3rd (1984) The regional distribution and cellular localization of iron in the rat brain. Neuroscience 11:595-603.
Hwang IK, Eum WS, Yoo KY, Cho JH, Kim DW, Choi SH, Kang TC, Kwon OS, Kang JH, Choi SY, Won MH (2005) Copper chaperone for Cu,Zn-SOD supplement potentiates the Cu, Zn-SOD function of neuroprotective effects against ischemic neuronal damage in the gerbil hippocampus. Free Radic Biol Med 39:392-402.
Ishimaru H, Ishikawa K, Ohe Y, Takahashi A, Tatemoto K, Maruyama Y (1996) Activation of iron handling system within the gerbil hippocampus after cerebral ischemia. Brain Res 726:23-30.
Jung HY, Yim HS, Yoo DY, Kim JW, Chung JY, Seong JK, Yoon YS, Kim DW, Hwang IK (2016) Postnatal changes in glucose transporter 3 expression in the dentate gyrus of the C57BL/6 mouse model. Lab Anim Res 32:1-7.
Kil HY, Zhang J, Piantadosi CA (1996) Brain temperature alters hydroxyl radical production during cerebral ischemia/reperfusion in rats. J Cereb Blood Flow Metab 16:100-106.
Kondo Y, Ogawa N, Asanuma M, Ota Z, Mori A (1995) Regional differences in late-onset iron deposition, ferritin, transferrin, astrocyte proliferation, and microglial activation after transient forebrain ischemia in rat brain. J Cereb Blood Flow Metab 15:216-226.
Li Y, He Y, Guan Q, Liu W, Han H, Nie Z (2012) Disrupted iron metabolism and ensuing oxidative stress may mediate cognitive dysfunction induced by chronic cerebral hypoperfusion. Biol Trace Elem Res 150:242-248.
Lin F, Girotti AW (1997) Elevated ferritin production, iron containment, and oxidant resistance in hemin-treated leukemia cells. Arch Biochem Biophys 346:131-141.
Lipscomb DC, Gorman LG, Traystman RJ, Hurn PD (1998) Low molecular weight iron in cerebral ischemic acidosis in vivo. Stroke 29:487-492.
Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79:1431-1568.
Loskota WA, Lomax P, Verity MA (1974) A stereotaxic atlas of the Mongolian gerbil brain (Meriones unguiculatus). Ann Arbor: Ann Arbor Science Publishers Inc.
Lozoff B, Georgieff MK (2006) Iron deficiency and brain development. Semin Pediatr Neurol 13:158-165.
Moos T, Rosengren Nielsen T, Skjørringe T, Morgan EH (2007) Iron trafficking inside the brain. J Neurochem 103:1730-1740.
Oberle S, Polte T, Abate A, Podhaisky HP, Schröder H (1998) Aspirin increases ferritin synthesis in endothelial cells: a novel antioxidant pathway. Circ Res 82:1016-1020.
Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR (2009) Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci 29:8828-8838.
Snyder AM, Connor JR (2009) Iron, the substantia nigra and related neurological disorders. Biochim Biophys Acta 1790:606-614.
Sorond FA, Ratan RR (2000) Ironing-out mechanisms of neuronal injury under hypoxic-ischemic conditions and potential role of iron chelators as neuroprotective agents. Antioxid Redox Signal 2:421-436.
Sugaya T, Kitani Y (1993) Isoflurane reduces microtubule-associated protein 2 degradation compared with halothane during forebrain ischaemia in the rat. Br J Anaesth 71:247-252.
Yang L, Zhang B, Yin L, Cai B, Shan H, Zhang L, Lu Y, Bi Z (2011) Tanshinone IIA prevented brain iron dyshomeostasis in cerebral ischemic rats. Cell Physiol Biochem 27:23-30.
Yoo DY, Kim W, Nam SM, Chung JY, Choi JH, Yoon YS, Won MH, Hwang IK (2012a) Chronic effects of pyridoxine in the gerbil hippocampal CA1 region after transient forebrain ischemia. Neurochem Res 37:1011-1018.
Yoo DY, Shin BN, Kim IH, Kim DW, Yoo KY, Kim W, Lee CH, Choi JH, Yoon YS, Choi SY, Won MH, Hwang IK (2012b) Effects of sensitive to apoptosis gene protein on cell proliferation, neuroblast differentiation, and oxidative stress in the mouse dentate gyrus. Neurochem Res 37:495-502.
Yoo KY, Hwang IK, Kang IJ, Kang TC, Lee HJ, Kang HY, Lee HY, Oh YS, Won MH (2007) Age-dependent changes in iron deposition in the gerbil hippocampus. Exp Anim 56:21-28.
Youdim MB, Ben-Shachar D, Riederer P (1991) Iron in brain function and dysfunction with emphasis on Parkinson's disease. Eur Neurol 31 Suppl 1:34-40.
Youdim MB, Stephenson G, Ben Shachar D (2004) Ironing iron out in Parkinson's disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann N Y Acad Sci 1012:306-325.
Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5:863-873.
Copyedited by Hwang IK, Li CH, Song LP, Zhao M
10.4103/1673-5374.184490
How to cite this article: Yoo DY, Yoo KY, Park JH, Kwon HJ, Jung HY, Kim JW, Choi GM, Moon SM, Kim DW, Yoon YS, Won MH, Hwang IK (2016) Time- and cell-type specific changes in iron, ferritin, and transferrin in the gerbil hippocampal CA1 region after transient forebrain ischemia. Neural Regen Res 11(6)∶924-930.
Funding: This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, No. 2015R1D1A1A01059980. This study was also partially supported by the Research Institute for Veterinary Science, Seoul National University.
*Correspondence to: In Koo Hwang, D.V.M., Ph.D., vetmed2@snu.ac.kr.
杂志排行

中国神经再生研究(英文版)的其它文章
- Bone marrow mesenchymal stem cell therapy in ischemic stroke: mechanisms of action and treatment optimization strategies
- Optic radiation injury in a patient with intraventricular hemorrhage: a diffusion tensor tractography study
- Synergetic effects of ciliary neurotrophic factor and olfactory ensheathing cells on optic nerve reparation (complete translation)
- miR-148b-3p promotes migration of Schwann cells by targeting cullin-associated and neddylationdissociated 1
- Transplantation of human adipose tissue-derived stem cells for repair of injured spiral ganglion neurons in deaf guinea pigs
- Indirubin-3′-monoxime suppresses amyloid-betainduced apoptosis by inhibiting tau hyperphosphorylation