Iron metabolism, oxidative stress, and neonatal brain injury
2016-12-02QingLu,StephenM.Black
PERSPECTIVE
Iron metabolism, oxidative stress, and neonatal brain injury
The brain injury associated with neonatal hypoxia ischemia (HI) is a major contributor to neonatal mortality and neurodevelopment retardation. Approximately 30—40% of infants with brain injury will die and 20—40% of survivors will develop significant neurological disorders and lifelong disability, such as cerebral palsy, seizures, visual impairment, mental retardation, learning impairment and epilepsy. Preterm birth is a high risk factor for neonatal brain injury. In the U.S., nearly 12% of babies are born preterm, and care of preterm infants accounts for more than half of pediatric health care dollars spent (Juul and Ferriero, 2014). A greater understanding of the complex mechanisms that result in neonatal hypoxia ischemic brain injury is the key to developing new protective therapeutics.
Both excitotoxicity and inflammation play important roles in the neuronal death associated with hypoxia-ischemic brain. Excessive Ca2+influx induced by glutamate receptor activation, activates proteases, phospholipases, endonucleases, and inhibits protein synthesis in neuron (Lipton and Rosenberg, 1994; Jin et al., 2010). The excessive Ca2+influx also activates neuronal nitric oxide synthase to generate nitric oxide (NO). NO has been considered a mediator of cerebral ischemic brain injury since it was observed that there is a burst of NO production in the rat brain at the initiation of the cerebral ischemia (Kader et al., 1993). However, the role of NO in the developing brain remains incompletely understood. It has been suggested that NO derived from endothelial (e)NOS is protective to the ischemic brain (Cimino et al., 2005), while delayed NO produced by inducible (i)NOS causes prolonged damage (Iadecola et al., 1997). But the most likely source of the early burst of NO is via the calcium mediated activation of neuronal (n)NOS (Huang et al., 1994). Previously, we have shown that nNOS is detected in the developing forebrain in the 10-day-old mouse embryo (E10). From E14 to E18, the highest level of expression was in the cortical plate neurons. However, this expression diminished with time; in the adult there were only a few nNOS-positive neurons in the deep layers of the cortex. Thus, nNOS expression in the developing brain, correlates with regions of selective vulnerability to hypoxic-ischemic injury, and supports a role for NO in hypoxic-ischemic injury in the developing brain (Black et al., 1995). In adult nNOS—/—mice, brain infarct volumes are significantly less after permanent middle cerebral artery occlusion (Huang et al., 1994). Similarly, neonatal nNOS—/—mice have less severe injury when exposed to HI (Ferriero et al., 1996). These studies have helped to clarify that nNOS derived NO contributes to the development of HI brain injury. But it is still not clear the mechanisms by which NO is involved in the HI brain injury. Some have suggested that nNOS-derived NO activates the p38MAPK pathway that leads to neuronal death (Li et al., 2013). This is supported by our previous studies that the activation of p38MAPK appears to be an initiator event which in turn results in an increase in oxidative stress through its ability to phosphorylate and activate the p47phoxsubunit of NADPH oxidase resulting in an increase in superoxide generation (Lu et al., 2012). Reactive oxygen species (ROS), such as superoxide, can damage neuronal cells through lipid peroxidation, protein oxidation, and DNA damage. Intracellular ROS are mainly processed by a highly complex and integrated antioxidant defense system composed of the enzymes CuZnSOD, MnSOD, catalase, GPx, and glutathione reductase, as well as non-enzymatic substances such as vitamin A, C, and E and low molecular weight molecules, including reduced GSH (Jiang et al., 2004). The ability to scavenge ROS is especially important for the protection of developing neonatal brain as it has high concentration of lipids, relatively high oxygen consumption, and low antioxidant defense systems, both enzymatic and non-enzymatic. Indeed our prior studies have shown that peak antioxidant defense protein levels, especially GPx, do not occur until later developmental ages (Khan and Black, 2003). However, the potential for antioxidant enzymes to exert neuroprotection is complex. For example, neonatal SOD1 over-expressing mice have increased brain injury. This appears to be due to the transgenic neonatal mice SOD1 is capable dismutate superoxide into hydrogen peroxide (H2O2), but without the compensatory capacity of catalase or GPx, H2O2accumulates to toxic levels. This enzymatic imbalance may be responsible for the increased free radical induced damage in the neonatal brain (Fullerton et al., 1998). As brain catalase levels are low compared to other tissues, we have suggested that GPx may be the important anti-oxidant enzyme in protecting the neonatal brain against HI injury, by showing that H2O2levels are significantly increased by oxygen glucose deprivation (OGD) in hippocampal slice cultures and by HI in the neonatal rat brain. Further, using the AAV gene delivery of GPx1, to scavenge H2O2, significantly attenuates the neuronal HI injury (Lu et al., 2012). More recently, we have been able to link increased generation of NO and H2O2with the neuronal injury associated with neonatal HI via changes in cellular iron metabolism. As there is rapid growth in early infancy the requirement for iron increases markedly. As such, the infant has a total body iron content of approximately 80 mg/kg, much higher than that in the adult. The requirement for iron in the brain is particularly high during neurodevelopment, primarily because iron is an enzymatic cofactor in myelinogenesis (Hare et al., 2015). Iron and transferrin levels have been reported to be high in cerebrospinal fluid, especially in perinatal brains (Erikson et al., 1997). Prior work has also shown that, in the neonatal HI rat brain, iron deposition increases rapidly in regions of ischemic injury, suggesting that HI induces a rapid accumulation of free iron in the brain (Palmer et al., 1999), while iron accumulation in oligodendrocytes induces their apoptosis (Rathnasamy et al., 2015). Further, we have recently identified a link between the increased NO generation associated with neonatal HI and the increase in iron deposition (Lu et al., 2015). The main mechanism for the regulation of intracellular iron homeostasis relies on a coordinated control of transferrin receptor (TfR)-mediated iron uptake and ferritin-mediated iron sequestration. The expression of TfR and ferritin is controlled by a process involving mRNA-protein interactions. The mRNA's of both TfR and ferritin contain structural motifs known as “iron response elements”(IREs), which can be bound by iron regulatory protein 1 (IRP-1) in response to intracellular iron levels. IRP-1 is activated when NO directly attacks the Fe—S cluster of aconitase, inducing its disassembly and switching the enzyme to IRP-1 (Pantopoulos and Hentze, 1995). This results in IRP-1 binding to IREs in TFR and ferritin and the subsequent attenuation of the degradation of the TfR mRNA and the simulataneous decrease in the translation of the ferritin mRNA. This produces a simultaneous increase in iron uptake and a decrease in iron sequestration. We have recently shown that a similar mechanism is responsible for the increase in iron deposition in the neonatal brain exposed to HI and that both NOS inhibition and iron scavenging decreased iron accumulation and reduced neonatal brain injury. Further, we identified that the mechanism by which the increase in free iron caused neuronal cell death is via its reaction with H2O2viaFenton chemistry that results in the generation of the highly reactive hydroxyl radical (·OH) (Lu et al., 2015) (Figure 1).
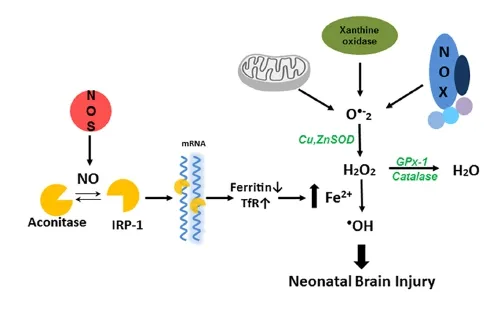
Figure 1 Nitric oxide dependent hydroxyl radical generation in neonatal hypoxia ischemia brain injury.
Thus, in conclusion there is a complex interplay between NO, oxidative stress and iron metabolism that underlies the neuronal cell death associated with neonatal HI. We speculate that therapies targeted at reducing H2O2levels or preserving the Fe—S cluster of aconitase may be effective in reducing the morbidity and mortality associated with neonatal HI. However, it is worth noting that recent studies in Alzheimer's disease have identified a role for Ca2+-sensing receptors (CaSRs) and CaSR antagonism using calcilytics suppresses the pathologic signaling associated with Aβ-CaSR (Dal Pra et al., 2015). Thus, CaSR antagonism may also be a therapeutic approach worth investigating in neonatal HI brain injury either alone or in combination with therapies that reduce H2O2or preserve aconitase activity.
This work was supported in part by National Institutes of Health grants (HL60190, HL67841, and P01HL0101902).
The authors wish to thank Valerie Harris for her exceptional technical work with the rat neonatal HI model.
Qing Lu, Stephen M. Black*
Department of Medicine, University of Arizona, Tucson, AZ, USA
*Correspondence to: Stephen M. Black, Ph.D., steveblack@email.arizona.edu.
Accepted: 2016-03-09
Black SM, Bedolli MA, Martinez S, Bristow JD, Ferriero DM, Soifer SJ (1995) Expression of neuronal nitric oxide synthase corresponds to regions of selective vulnerability to hypoxia-ischaemia in the developing rat brain. Neurobiol Dis 2:145-155.
Dal Pra I, Chiarini A, Armato U (2015) Antagonizing amyloid-beta/calcium-sensing receptor signaling in human astrocytes and neurons: a key to halt Alzheimer's disease progression? Neural Regen Res 10:213-218.
Ferriero DM, Holtzman DM, Black SM, Sheldon RA (1996) Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis 3:64-71.
Fullerton HJ, Ditelberg JS, Chen SF, Sarco DP, Chan PH, Epstein CJ, Ferriero DM (1998) Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol 44:357-364.
Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA (1994) Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 265:1883-1885.
Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME (1997) Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci 17:9157-9164.
Jiang X, Mu D, Manabat C, Koshy AA, Christen S, Tauber MG, Vexler ZS, Ferriero DM (2004) Differential vulnerability of immature murine neurons to oxygen-glucose deprivation. Exp Neurol 190:224-232.
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87:779-789.
Juul SE, Ferriero DM (2014) Pharmacologic neuroprotective strategies in neonatal brain injury. Clin Perinatol 41:119-131.
Kader A, Frazzini VI, Solomon RA, Trifiletti RR (1993) Nitric oxide production during focal cerebral ischemia in rats. Stroke 24:1709-1716.
Khan JY, Black SM (2003) Developmental changes in murine brain antioxidant enzymes. Pediatr Res 54:77-82.
Li LL, Ginet V, Liu X, Vergun O, Tuittila M, Mathieu M, Bonny C, Puyal J, Truttmann AC, Courtney MJ (2013) The nNOS-p38MAPK pathway is mediated by NOS1AP during neuronal death. J Neurosci 33:8185-8201.
Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613-622.
Lu Q, Harris VA, Rafikov R, Sun X, Kumar S, Black SM (2015) Nitric oxide induces hypoxia ischemic injury in the neonatal brain via the disruption of neuronal iron metabolism. Redox biology 6:112-121.
Lu Q, Wainwright MS, Harris VA, Aggarwal S, Hou Y, Rau T, Poulsen DJ, Black SM (2012) Increased NADPH oxidase-derived superoxide is involved in the neuronal cell death induced by hypoxia-ischemia in neonatal hippocampal slice cultures. Free Radic Biol Med 53:1139-1151.
Palmer C, Menzies SL, Roberts RL, Pavlick G, Connor JR (1999) Changes in iron histochemistry after hypoxic-ischemic brain injury in the neonatal rat. J Neurosci Res 56:60-71.
Pantopoulos K, Hentze MW (1995) Nitric oxide signaling to iron-regulatory protein: direct control of ferritin mRNA translation and transferrin receptor mRNA stability in transfected fibroblasts. Proc Natl Acad Sci U S A 92:1267-1271.
Rathnasamy G, Murugan M, Ling EA, Kaur C (2015) Hypoxia-induced iron accumulation in oligodendrocytes mediates apoptosis by eliciting endoplasmic reticulum stress. Mol Neurobiol doi:10.1007/s12035-015-9389-6.
10.4103/1673-5374.182691 http∶//www.nrronline.org/
How to cite this article: Lu Q, Black SM (2016) Iron metabolism, oxidative stress, and neonatal brain injury. Neural Regen Res 11(5):725-726.
杂志排行

中国神经再生研究(英文版)的其它文章
- Possible application of apolipoprotein E-containing lipoproteins and polyunsaturated fatty acids in neural regeneration
- Recovery of injured fornical crura following neurosurgical operation of a brain tumor: a case report
- Antibody-based neuronal and axonal delivery vectors for targeted ligand delivery
- Coordination of the axonal cytoskeleton during the emergence of axon collateral branches
- Alzheimer's disease: the silver tsunami of the 21stcentury
- Clinical trial perspective for adult and juvenile Huntington's disease using genetically-engineered mesenchymal stem cells