Vascular endothelial growth factor: an attractive target in the treatment of hypoxic/ischemic brain injury
2016-12-01HuiGuoHuiZhouJieLuYiQuDanYuYuTong
Hui Guo, Hui Zhou, Jie Lu, Yi Qu Dan Yu, Yu Tong
1 Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, Sichuan Province, China
2 Key Laboratory of Obstetric & Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, Sichuan Province, China
3 Department of Medical Cosmetology, Chengdu Second People’s Hospital, Chengdu, Sichuan Province, China
REVIEW
Vascular endothelial growth factor: an attractive target in the treatment of hypoxic/ischemic brain injury
Hui Guo1,2,#, Hui Zhou1,2,#, Jie Lu3,#, Yi Qu1,2, Dan Yu1,2,*, Yu Tong1,2,*
1 Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, Sichuan Province, China
2 Key Laboratory of Obstetric & Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, Sichuan Province, China
3 Department of Medical Cosmetology, Chengdu Second People’s Hospital, Chengdu, Sichuan Province, China
Cerebral hypoxia or ischemia results in cell death and cerebral edema, as well as other cellular reactions such as angiogenesis and the reestablishment of functional microvasculature to promote recovery from brain injury. Vascular endothelial growth factor is expressed in the central nervous system after hypoxic/ ischemic brain injury, and is involved in the process of brain repair via the regulation of angiogenesis, neurogenesis, neurite outgrowth, and cerebral edema, which all require vascular endothelial growth factor signaling. In this review, we focus on the role of the vascular endothelial growth factor signaling pathway in the response to hypoxic/ischemic brain injury, and discuss potential therapeutic interventions.
nerve regeneration; VEGF; VEGFR; HIF1; PI3K/Akt pathway; Akt/eNOS pathway; JAK/STAT; Src-SSeCKS pathway; hypoxic/ischemic brain injury; neural regeneration
# These authors contributed equally to this work.
orcid: 0000-0002-1744-6020 (Yu Tong) 0000-0002-7785-9951 (Dan Yu)
http://www.nrronline.org/
Accepted: 2015-10-12
Introduction
Cerebral hypoxia leads to necrosis and apoptosis, in addition to other cellular reactions, such as angiogenesis, which promote recovery from brain injury (Bhattacharya et al. 2013). The vascular endothelial growth factor (VEGF) family comprises the trophic factors VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor, and stimulates the growth of new blood vessels (Holmes et al., 2007). VEGF is expressed in the central nervous system (CNS) after injury (Dore-Duffy et al., 2007; Chaitanya et al., 2013; Leonard and Gulati, 2013) and is an important regulator of vascular leakage in the brain. Hypoxia induces the expression of VEGF, leading to the formation of cerebral edema (Bauer et al., 2010; Baburamani et al., 2013).
VEGF can reduce the damage caused by hypoxia/ischemia in a number of ways (Shimotake et al., 2010; Zhao et al., 2011; Dzietko et al., 2013). During cerebral ischemia, VEGF promotes neurogenesis (van Rooijen et al., 2010; Moriyama et al., 2013; Rosell et al., 2013), neurite outgrowth (Cesca et al., 2012), and the survival of newborn neuronal precursors. It also has an important role in increasing the size of the subventricular zone after injury (Gotts and Chesselet, 2005). Furthermore, inhibition of VEGF expression after injury may exacerbate neuronal and glial damage (Skold et al., 2006). However, increased endogenous VEGF interacts with its receptors on ischemic vessels, and contributes to the disruption of the blood-brain barrier and subsequent leakage (Zhang et al., 2011). Therefore, understanding the relevant signaling pathway of VEGF in response to hypoxia/ ischemia, and devising ways to modulate it, is essential for the successful treatment of hypoxic/ischemic brain injury. In this review, we provide an overview of the VEGF signaling pathway and discuss its role in hypoxic/ischemic brain injury.
VEGF and Its Receptors (VEGFRs)
VEGF is an endothelial cell-specific mitogen and a secreted dimeric protein, and as such can induce angiogenesis in a variety of ways (Dzietko et al., 2013; Morgan et al., 2007; Holzer et al., 2013). The role of VEGF in angiogenesis is crucial for the development and regeneration/restoration of tissue, as well as for tumor formation (Morgan et al., 2007; Dzietko et al., 2013; Holzer et al., 2013). Rodent models of hypoxia/ischemia have demonstrated that angiogenesisprovides the right neurovascular microenvironment for neuronal remodeling (Arai et al., 2009; Xiong et al., 2011). Lin et al. (2003) first found that VEGF promotes the outgrowth of nerve fibers on cultured major pelvic ganglia in vitro. A large number of studies have suggested that activation of the VEGF★VEGFR signaling pathway is beneficial to neurobehavioral recovery and neurovascular remodeling after hypoxic/ischemic brain injury (Shimotake et al., 2010; Zhao et al., 2011; Dzietko et al., 2013).
In most mammalian tissues, VEGF165 is the most common isoform of VEGF, existing as a heparin-binding homodimeric glycoprotein of approximately 45 kDa (Holzer et al., 2013). VEGF regulates physiological and pathological angiogenesis by binding to and activating the tyrosine kinase receptors VEGFR-1 (Flt-1) and VEGFR-2 (Flk-1) (Ho et al., 2012). In vascular endothelial cells, VEGF binds to VEGFR-1 and (predominantly) VEGFR-2, and stimulates angiogenesis in the periphery by triggering mitotic and migratory processes (Shibuya and Claesson-Welsh, 2006). VEGFR-2 is considered the major receptor for VEGF-mediated activities (Zachary and Gliki, 2001). VEGFR-1 binds to endogenous VEGF and transmits the proliferation signal for astrocytes and the vasculature (Shih et al., 2003; Krum et al., 2008; Sato et al., 2011). VEGFR-1 is upregulated almost exclusively in reactive astrocytes (Krum and Khaibullina, 2003; Khaibullina et al., 2004), while VEGFR-2 is upregulated in neurons. Furthermore, it has been shown that VEGF stimulates axonal outgrowth by binding to VEGFR-2. In situ hybridization and immunocytochemistry in adult mice revealed that VEGF promotes axonal outgrowth from dorsal root ganglia, and that the VEGFR-2 inhibitor SU5416 prevented this process (Sondel et al., 1999; Olbrich et al., 2012). These findings provide sound evidence that VEGF is necessary for the regeneration of peripheral nerves.
VEGF and Hypoxia Inducible Factor (HIF)
HIFs are important regulators of the transcriptional response to oxygen deprivation. In the adult hypoxic brain, the nuclear protein complex HIF-1 is the most ubiquitously expressed member of the HIF family. It is the best-characterized transcription regulator of VEGF, and binds to the consensus sequence in target gene promoters. HIF-1 is a heterodimer composed of an alpha and a beta subunit. The beta subunit has been identified as the aryl hydrocarbon receptor nuclear translocator. Hypoxia induces HIF-1 expression (Josko and Mazurek, 2004; Dery et al., 2005). Under normoxic conditions, HIF-1α is rapidly degraded by the ubiquitin-proteosome system, but remains stable during hypoxia. Conversely, HIF-1α is stable under normoxic conditions. The expression of HIF-1α is increased in different cell types during hypoxia-induced CNS injury (Jin et al., 2000). Furthermore, Marti et al. (2000) revealed that HIF-1 and VEGF mRNA are coexpressed in a mouse model of focal ischemia, and that the number of newly formed vessels is increased at the marginal zone of the cerebral infarction. The same group also analyzed the expression of VEGF and VEGFRs in hypoxic cells, observing a significant increase both in VEGF in the ischemic region and in VEGFRs at the border. They further found that expression of HIF-1 was also increased in the ischemic region. These results strongly suggest that the HIF-1-VEGF-VEGFR signaling pathway may be involved in the growth of new vessels after cerebral ischemic injury.
In another study, Nordal et al. (2004) used immunohistochemistry and in situ hybridization to detect the expression of the HIF-1α subunit and VEGF in the irradiated rat spinal cord. HIF-1α expression was observed in glial cells expressing VEGF (Sondell et al., 2000), and VEGF expression correlated with HIF-1α expression. A number of HIF-1α-mediated regulators of genes such as VEGF and erythropoietin may be relevant in CNS injury responses (Mu et al., 2003). In the ischemic or hypoxic brain, astrocytes are one of the main sources of erythropoietin. The pathway by which HIF-1α mediates the transcriptional activation of erythropoietin expression may promote the survival of neurons during hypoxia via an astrocytic paracrine-dependent mechanism (Fandrey, 2004). By activating the phosphatidylinositol-3-kinase (PI3K)-Akt and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathways, erythropoietin increases the secretion of VEGF in neural stem cells (Xiong et al., 2011). Upregulation of VEGF increases vascular permeability and interstitial fluid pressure, and reduces perfusion and edema. Although the precise mechanism by which VEGF increases permeability remains unclear, it may involve action on tight junction proteins or adhesion molecules (Radisavljevic et al., 2000; Fischer et al., 2002). Interrupting this secondary cycle of damage caused by VEGF upregulation may improve neuroprotective strategies against CNS radiation injury. Above all, VEGF may be involved in hypoxic/ischemic brain injury via the HIF-erythropoietin-PI3K-Akt and ERK1/2-VEGF pathways.
VEGF and the VEGFR-2-Akt-endothelial nitric oxide synthase (eNOS) pathway
raumatic brain injury (TBI) remains one of the main causes of serious, long-term disability. One of the most prominent pathophysiological changes after TBI is ischemia and hypoxia in the lesion boundary area, and the volume of ischemic tissue in early focal cerebral ischemia after TBI correlates with neurological outcome (Coles et al., 2004). Following TBI, a substantial increase in angiogenesis occurs, which may provide oxygen and nutrition for cerebral reconstruction (Morgan et al., 2007). TBI-induced angiogenesis and functional recovery in the lesion boundary zone and hippocampus are improved by simvastatin, an effect which may be mediated by activation of the VEGFR-2-Akt-eNOS signaling pathway (Wu et al., 2011). In vitro, simvastatin can stimulate endothelial cell tube formation after oxygen-glucose deprivation. Simvastatin can also augment the expression of VEGFR-2 in brain tissue and cultured rat microvascular endothelial cells, and this effect may be related to simvastatin-induced activation of Akt. Furthermore, simvastatin can also induce Akt-dependent eNOS phosphorylation in vivo and in vitro (Wu et al., 2011).
Many of the downstream angiogenic effects of VEGF, suchas microvascular permeability and endothelial cell proliferation, migration and survival, are mediated by VEGFR-2 (Hicklin and Ellis, 2005). On the surface of endothelial cells, VEGF activates intracellular tyrosine kinases by binding to VEGFR-2, which triggers multiple downstream signals to stimulate angiogenesis. Among these, Akt-dependent eNOS phosphorylation is essential for angiogenesis (Kureishi et al., 2000). Phosphorylation of the protein kinase Akt plays a crucial role in multiple cellular and physiologic effects (Parcellier et al., 2008). The pro-survival effects of Akt include anti-apoptosis, angiogenesis, and neuroprotection after brain injury (Kilic et al., 2006; Shein et al., 2007). In a TBI study (Thau-Zuchman et al., 2010), the effects of VEGF on brain recovery and function were examined by infusing ectogenic VEGF into the lateral ventricles of mice for 7 days after TBI. VEGF had multiple effects, including promotion of neurogenesis and angiogenesis, neuroprotection, and improvement of functional recovery by mediating phospho-Akt signaling (Thau-Zuchman et al., 2010). eNOS is a downstream mediator of VEGFR-2 and is critical for angiogenesis (Fischer et al., 2002). eNOS mediates vasodilation after hypoxic/ ischemic episodes by increasing blood flow (Bolanos and Almeida, 1999). Nitric oxide is synthesized by eNOS and is an essential component of the pathological and physiological response to hypoxia/ischemia (Kaur and Ling, 2008). Simvastatin administration can activate Akt-GSK-3 and enhance phosphorylation of eNOS in the TBI model (Thau-Zuchman et al., 2010), and phospho-eNOS in turn induces a series of downstream effects, such as angiogenesis and recruitment of mural cells to immature angiogenic sprouts (Kashiwagi et al., 2005).
VEGF and the VEGFR-2-PI3K-Akt pathway
The pro-angiogenic effects of VEGF are thought to be attributed to VEGFR-2, and the protective effect of VEGF on cerebral cortical neurons may involve VEGFR-2 dimerization to form a receptor complex with neuropilin-1 (Sato et al., 2011). Class Ia PI3K and its downstream effector Akt are enabled by the activation of VEGFR-2 (Koch et al., 2011). The PI3K-Akt pathway is crucial for many VEGF-dependent effects, including cell survival and migration, and vasopermeability (Olsson et al., 2006). The VEGF-PI3K-Akt pathway is not only involved in endothelial permeability in vitro (Pedram et al., 2002), but is also attributed to neuroprotection and blood-brain barrier permeability in a mouse model of focal cerebral ischemia. Furthermore, these effects are dependent on VEGFR-2 (Hicklin and Ellis, 2005). This pathway may contribute to the maintenance of mitochondrial function under conditions of tissue oxygen deficit. Akt is activated by phosphorylation of the Bcl-2-associated death promoter, which increases the removal of Bcl-xL from mitochondria, blocks the formation of the mitochondrial permeability transition pore, and maintains the mitochondrial membrane potential. In addition, Bcl-xL depresses the activity of caspases 9 and 3 by blocking the release of cytochrome c from injured mitochondria, thereby restraining DNA cleavage (Cheng et al., 2010; Wu et al., 2011).
The specific mechanism by which VEGF-VEGFR-2 activates PI3K-Akt is still unclear, but a recent report suggested that the receptor tyrosine kinase Axl may be responsible for VEGF-A-dependent activation of PI3K/Akt (Ruan and Kazlauskas, 2012).
VEGF and the JAK-STAT pathway
The Janus kinase (JAK) family comprises four non-receptor tyrosine kinases, JAK1, JAK2, JAK3 and TYK2. The first three are widely expressed in various tissues and cells, but TYK3 exists only in the bone marrow and lymph system. Signal transducer and activator of transcription (STAT) is the substrate for JAK. The STAT family comprises seven latent cytoplasmic transcription factors that are involved in signal transduction mediated by cytokines and growth factors. The JAK-STAT pathway is downstream of the cytokine receptors. Activation of these cytokine receptors initiates JAK phosphorylation and activation, which in turn phosphorylates STAT. Following tyrosine phosphorylation, STAT proteins dimerize through the nuclear membrane into the nucleus, where they combine with genomic regulatory sequences and enhance the transcription of related genes (Lai and Johnson, 2010).
In mammals, the JAK-STAT pathway is considered to be the major signaling mechanism for a number of growth factors and cytokines (Ihle and Kerr, 1995), and mediates a wide variety of biological functions in the CNS including the regeneration of peripheral nerves (Bella et al., 2006; Lin et al., 2006; Xu et al., 2009), and may also be involved in axon regeneration and in the proliferation and migration of Schwann cells after sciatic nerve injury (Xu et al., 2009).
To date, most studies on the interaction between VEGF and the JAK-STAT pathway have focused on tumors (Roorda et al., 2010) and cellular invasiveness. Whether VEGF can directly activate the JAK-STAT pathway to promote neurite outgrowth has not been examined. VEGF may achieve this by promoting angiogenesis or by binding with JAK-STAT directly, similarly to combining with neurotrophic factors. However, activation of STAT3 can increase VEGF expression, which indicates that another signaling pathway may be involved (Wang et al., 2010). VEGF expression can be induced by latent membrane protein 1 via both the JAKSTAT and mitogen-activated protein kinase (MAPK)-ERK pathways (Wang et al., 2010). Furthermore, the increased expression of VEGF induced by elevated phosphorylation of STAT3 after nerve injury may further sensitize the JAKSTAT pathway (Bella., 2007). Therefore, VEGF may interact directly or indirectly with the JAK-STAT pathway. Further understanding of the interaction between VEGF and the JAK-STAT pathway will be beneficial to develop new therapies for neuronal recovery.
VEGF and the Src-SSeCKS pathway
The non-receptor tyrosine kinase Src is another protein considered to be associated with angiogenesis (Theus et al., 2006; Tang et al., 2007). The activity of Src kinase increases significantly during transient global ischemia (Schlessinger,2000) and this effect is associated with increased vascular permeability mediated by VEGF (Paul et al., 2001; Zan et al., 2014; He et al., 2015). Mice lacking the Src subtype pp60c-Src are resistant to this increase in VEGF-induced vascular permeability and have smaller infarct volumes after stroke. However, mice lacking pp59c-Fyn, another member of the Src family, do not show these effects (Paul et al., 2001). VEGF induces endothelial activation and vascular leak mainly via VEGFR-2 (Mason et al., 2004) and Src (Eliceiri et al., 1999). Src-suppressed C kinase substrate (SSeCKS) is widely expressed in astrocyte-like, neuron-like and endothelial-like cells (Zan et al., 2011). Under ischemic conditions, Src and SSeCKS can regulate the expression of VEGF (Lee et al., 2003; Zan et al., 2011). Inhibition of Src decreases VEGF-induced vascular permeability and infarct volume (Bella et al., 2007) and alleviates brain edema and injury (Akiyama et al., 2004; Lennmyr et al., 2004). Accordingly, inhibitors of VEGF or Src kinases can reduce the edema and tissue injury following myocardial ischemia injury (Weis et al., 2004). The inhibition of SSeCKS by Src and its regulation of angiogenesis and vascular permeability may be achieved by regulating VEGF and tight junction proteins after ischemic brain injury. Src and SSeCKS may have opposing effects on angiogenesis and vascular permeability after focal cerebral ischemia, and angiogenic factors are involved in this process by serving as downstream mediators (Paul et al., 2001). Furthermore, modulation of angiogenesis and vascular leakage by the Src-SSeCKS pathway helps improve recovery after focal cerebral ischemia (Bai et al., 2014).
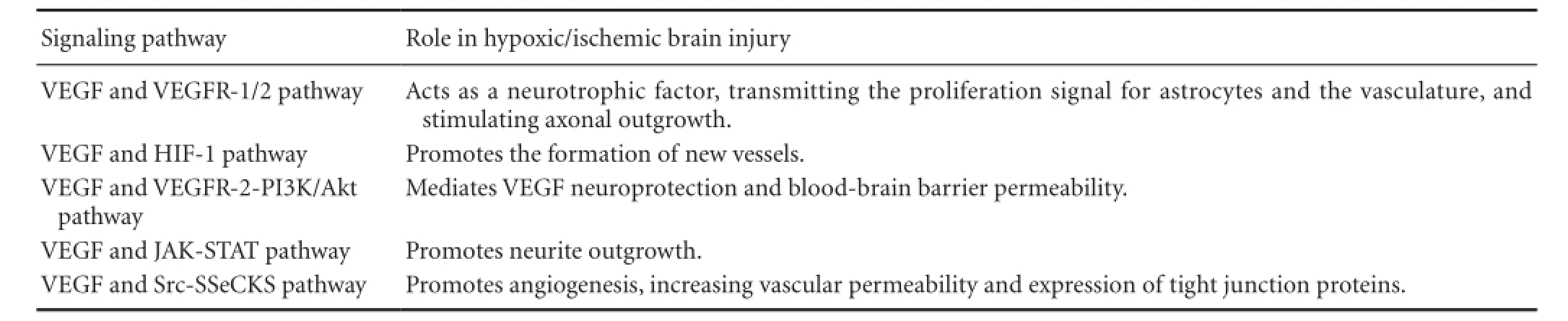
Table 1 VEGF pathways involved in hypoxic/ischemic brain injury
Conclusion
Hypoxic/ischemic brain injury causes severe brain damage, but the specific mechanisms underlying the pathophysiology of such injury, and preventive measures, remain unclear. Cerebral hypoxia/ischemia results in widespread responses at the systemic and cellular levels and regulates many physiological and pathological processes. The upregulation of VEGF is considered to be a crucial stimulus for these processes. As a hypoxia-induced angiogenic protein, VEGF plays a double-edged role in the central nervous system. Based on its trophic influence on neurons and vascular cells, it is a promising candidate for brain injury treatment. Accumulating evidence implicates VEGF in cerebral hypoxia/ischemia via the HIF-1, VEGF-R2-PI3K-Akt, VEGF-R2-Akt-eNOS, JAK-STAT, and Src-SSeCKS pathways (Table 1). Thus VEGF becomes an attractive target for the treatment of hypoxic/ ischemic brain injury. A variety of therapeutic strategies targeting VEGF are currently in the research pipeline, but most of them are in the experimental stages. Creatinine may be an effective treatment against cerebral hypoxia/ischemia, increasing the expression of VEGF and mediating neovascularization in the ischemic zone (Pedram et al., 2002); however, the downstream intracellular signaling pathways mediating these effects remain unclear. A better understanding of the VEGF signaling pathway will improve therapeutic advances for hypoxic/ischemic brain injury.
Author contributions: HZ and JL performed the literature search. DY and YQ performed the study selection. HG and YT wrote this paper. All authors approved the final version of this paper.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Akiyama C, Yuguchi T, Nishio M, Tomishima T, Fujinaka T, Taniguchi M, Nakajima Y, Kohmura E, Yoshimine T (2004) Src family kinase inhibitor PP1 reduces secondary damage after spinal cord compression in rats. J Neurotrauma 21:923-931.
Arai K, Jin G, Navaratna D, Lo EH (2009) Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J 276:4644-4652.
Baburamani AA, Castillo-Melendez M, Walker DW (2013) VEGF expression and microvascular responses to severe transient hypoxia in the fetal sheep brain. Pediatr Res 73:310-316.
Bai Y, Xu G, Xu M, Li Q, Qin X (2014) Inhibition of Src phosphorylation reduces damage to the blood-brain barrier following transient focal cerebral ischemia in rats. Int J Mol Med 34:1473-1482.
Bauer AT, Burgers HF, Rabie T, Marti HH (2010) Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J Cereb Blood Flow Metab 30:837-848.
Bella AJ, Lin G, Tantiwongse K, Garcia M, Lin CS, Brant W, Lue TF (2006) Brain-derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: part I. J Sex Med 3:815-820.
Bella AJ, Lin G, Garcia MM, Tantiwongse K, Brant WO, Lin CS, Lue TF (2007) Upregulation of penile brain-derived neurotrophic factor (BDNF) and activation of the JAK/STAT signalling pathway in the major pelvic ganglion of the rat after cavernous nerve transection. Eur Urol 52:574-580.
Bhattacharya P, Pandey AK, Paul S, Patnaik R, Yavagal DR (2013) Aquaporin-4 inhibition mediates piroxicam-induced neuroprotection against focal cerebral ischemia/reperfusion injury in rodents. PLoS One 8:e73481.
Bolanos JP, Almeida A (1999) Roles of nitric oxide in brain hypoxia-ischemia. Biochim Biophys Acta 1411:415-436.
Cesca F, Yabe A, Spencer-Dene B, Scholz-Starke J, Medrihan L, Maden CH, Gerhardt H, Orriss IR, Baldelli P, Al-Qatari M, Koltzenburg M, Adams RH, Benfenati F, Schiavo G (2012) Kidins220/ARMS mediates the integration of the neurotrophin and VEGF pathways in the vascular and nervous systems. Cell Death Differ 19:194-208.
Chaitanya GV, Cromer WE, Parker CP, Couraud PO, Romero IA, Weksler B, Mathis JM, Minagar A, Alexander JS (2013) A recombinant inhibitory isoform of vascular endothelial growth factor164/165 aggravates ischemic brain damage in a mouse model of focal cerebral ischemia. Am J Pathol 183:1010-1024.
Cheng XW, Kuzuya M, Kim W, Song H, Hu L, Inoue A, Nakamura K, Di Q, Sasaki T, Tsuzuki M, Shi GP, Okumura K, Murohara T (2010) Exercise training stimulates ischemia-induced neovascularization via phosphatidylinositol 3-kinase/Akt-dependent hypoxia-induced factor-1 alpha reactivation in mice of advanced age. Circulation 122:707-716.
Coles JP, Fryer TD, Smielewski P, Chatfield DA, Steiner LA, Johnston AJ, Downey SP, Williams GB, Aigbirhio F, Hutchinson PJ, Rice K, Carpenter TA, Clark JC, Pickard JD, Menon DK (2004) Incidence and mechanisms of cerebral ischemia in early clinical head injury. J Cereb Blood Flow Metab 24:202-211.
Dery MA, Michaud MD, Richard DE (2005) Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. Int J Biochem Cell Biol 37:535-540.
Dore-Duffy P, Wang X, Mehedi A, Kreipke CW, Rafols JA (2007) Differential expression of capillary VEGF isoforms following traumatic brain injury. Neurol Res 29:395-403.
Dzietko M, Derugin N, Wendland MF, Vexler ZS, Ferriero DM (2013) Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl Stroke Res 4:189-200.
Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA (1999) Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell 4:915-924.
Fandrey J (2004) Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am J Physiol Regul Integr Comp Physiol 286:R977-988.
Fischer S, Wobben M, Marti HH, Renz D, Schaper W (2002) Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res 63:70-80.
Gotts JE, Chesselet MF (2005) Vascular changes in the subventricular zone after distal cortical lesions. Exp Neurol 194:139-150.
He YX, Liu J, Guo B, Wang YX, Pan X, Li D, Tang T, Chen Y, Peng S, Bian Z, Liang Z, Zhang BT, Lu A, Zhang G (2015) Src inhibitor reduces permeability without disturbing vascularization and prevents bone destruction in steroid-associated osteonecrotic lesions in rabbits. Sci Rep 5:8856.
Hicklin DJ, Ellis LM (2005) Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 23:1011-1027.
Ho VC, Duan LJ, Cronin C, Liang BT, Fong GH (2012) Elevated vascular endothelial growth factor receptor-2 abundance contributes to increased angiogenesis in vascular endothelial growth factor receptor-1-deficient mice. Circulation 126:741-752.
Holmes K, Roberts OL, Thomas AM, Cross MJ (2007) Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 19:2003-2012.
Holzer LA, Cor A, Pfandlsteiner G, Holzer G (2013) Expression of VEGF, its receptors, and HIF-1alpha in Dupuytren’s disease. Acta Orthop 84:420-425.
Ihle JN, Kerr IM (1995) Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet 11:69-74.
Jin KL, Mao XO, Nagayama T, Goldsmith PC, Greenberg DA (2000) Induction of vascular endothelial growth factor and hypoxia-inducible factor-1alpha by global ischemia in rat brain. Neuroscience 99:577-585.
Josko J, Mazurek M (2004) Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Med Sci Monit 10:RA89-98.
Kashiwagi S, Izumi Y, Gohongi T, Demou ZN, Xu L, Huang PL, Buerk DG, Munn LL, Jain RK, Fukumura D (2005) NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J Clin Invest 115:1816-1827.
Kaur C, Ling EA (2008) Blood brain barrier in hypoxic-ischemic conditions. Curr Neurovasc Res 5:71-81.
Khaibullina AA, Rosenstein JM, Krum JM (2004) Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res Dev Brain Res 148:59-68.
Kilic E, Kilic U, Wang Y, Bassetti CL, Marti HH, Hermann DM (2006) The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J 20:1185-1187.
Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L (2011) Signal transduction by vascular endothelial growth factor receptors. Biochem J 437:169-183.
Krum JM, Khaibullina A (2003) Inhibition of endogenous VEGF impedes revascularization and astroglial proliferation: roles for VEGF in brain repair. Exp Neurol 181:241-257.
Krum JM, Mani N, Rosenstein JM (2008) Roles of the endogenous VEGF receptors flt-1 and flk-1 in astroglial and vascular remodeling after brain injury. Exp Neurol 212:108-117.
Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K (2000) The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 6:1004-1010.
Lai SY, Johnson FM (2010) Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist Updat 13:67-78.
Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, Kim YJ, Kim KW (2003) SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med 9:900-906.
Lennmyr F, Ericsson A, Gerwins P, Akterin S, Ahlstrom H, Terent A (2004) Src family kinase-inhibitor PP2 reduces focal ischemic brain injury. Acta Neurol Scand 110:175-179.
Leonard MG, Gulati A (2013) Endothelin B receptor agonist, IRL-1620, enhances angiogenesis and neurogenesis following cerebral ischemia in rats. Brain Res 1528:28-41.
Lin G, Bella AJ, Lue TF, Lin CS (2006) Brain-derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: part 2. J Sex Med 3:821-827.
Lin G, Chen KC, Hsieh PS, Yeh CH, Lue TF, Lin CS (2003) Neurotrophic effects of vascular endothelial growth factor and neurotrophins on cultured major pelvic ganglia. BJU Int 92:631-635.
Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W (2000) Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol 156:965-976.
Mason JC, Steinberg R, Lidington EA, Kinderlerer AR, Ohba M, Haskard DO (2004) Decay-accelerating factor induction on vascular endothelium by vascular endothelial growth factor (VEGF) is mediated via a VEGF receptor-2 (VEGF-R2)- and protein kinase C-alpha/epsilon (PKCalpha/epsilon)-dependent cytoprotective signaling pathway and is inhibited by cyclosporin A. J Biol Chem 279:41611-41618.
Morgan R, Kreipke CW, Roberts G, Bagchi M, Rafols JA (2007) Neovascularization following traumatic brain injury: possible evidence for both angiogenesis and vasculogenesis. Neurol Res 29:375-381.
Moriyama Y, Takagi N, Hashimura K, Itokawa C, Tanonaka K (2013) Intravenous injection of neural progenitor cells facilitates angiogenesis after cerebral ischemia. Brain Behav 3:43-53.
Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, Ferriero DM (2003) Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis 14:524-534.
Nordal RA, Nagy A, Pintilie M, Wong CS (2004) Hypoxia and hypoxia-inducible factor-1 target genes in central nervous system radiation injury: a role for vascular endothelial growth factor. Clin Cancer Res 10:3342-3353.
Olbrich L, Foehring D, Happel P, Brand-Saberi B, Theiss C (2012) Fast rearrangement of the neuronal growth cone’s actin cytoskeleton following VEGF stimulation. Histochem Cell Biol 139:431-445.
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L (2006) VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 7:359-371.
Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA (2008) PKB and the mitochondria: AKTing on apoptosis. Cell Signal 20:21-30.
Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA (2001) Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med 7:222-227.
Pedram A, Razandi M, Levin ER (2002) Deciphering vascular endothelial cell growth factor/vascular permeability factor signaling to vascular permeability. Inhibition by atrial natriuretic peptide. J Biol Chem 277:44385-44398.
Radisavljevic Z, Avraham H, Avraham S (2000) Vascular endothelial growth factor up-regulates ICAM-1 expression via the phosphatidylinositol 3 OH-kinase/AKT/Nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J Biol Chem 275:20770-20774.
Roorda BD, Ter Elst A, Scherpen FJ, Meeuwsen-de Boer TG, Kamps WA, de Bont ES (2010) VEGF-A promotes lymphoma tumour growth by activation of STAT proteins and inhibition of p27(KIP1) via paracrine mechanisms. Eur J Cancer 46:974-982.
Rosell A, Morancho A, Navarro-Sobrino M, Martinez-Saez E, Hernandez-Guillamon M, Lope-Piedrafita S, Barcelo V, Borras F, Penalba A, Garcia-Bonilla L, Montaner J (2013) Factors secreted by endothelial progenitor cells enhance neurorepair responses after cerebral ischemia in mice. PLoS One 8:e73244.
Ruan GX, Kazlauskas A (2012) Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J 31:1692-1703.
Sato W, Tanabe K, Kosugi T, Hudkins K, Lanaspa MA, Zhang L, Campbell-Thompson M, Li Q, Long DA, Alpers CE, Nakagawa T (2011) Selective stimulation of VEGFR2 accelerates progressive renal disease. Am J Pathol 179:155-166.
Schlessinger J (2000) New roles for Src kinases in control of cell survival and angiogenesis. Cell 100:293-296.
Shein NA, Tsenter J, Alexandrovich AG, Horowitz M, Shohami E (2007) Akt phosphorylation is required for heat acclimation-induced neuroprotection. J Neurochem 103:1523-1529.
Shibuya M, Claesson-Welsh L (2006) Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res 312:549-560.
Shih SC, Ju M, Liu N, Mo JR, Ney JJ, Smith LE (2003) Transforming growth factor beta1 induction of vascular endothelial growth factor receptor 1: mechanism of pericyte-induced vascular survival in vivo. Proc Natl Acad Sci U S A 100:15859-15864.
Shimotake J, Derugin N, Wendland M, Vexler ZS, Ferriero DM (2010) Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke 41:343-349.
Skold MK, Risling M, Holmin S (2006) Inhibition of vascular endothelial growth factor receptor 2 activity in experimental brain contusions aggravates injury outcome and leads to early increased neuronal and glial degeneration. Eur J Neurosci 23:21-34.
Sondell M, Lundborg G, Kanje M (1999) Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci 19:5731-5740.
Sondell M, Sundler F, Kanje M (2000) Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur J Neurosci 12:4243-4254.
Tang X, Feng Y, Ye K (2007) Src-family tyrosine kinase fyn phosphorylates phosphatidylinositol 3-kinase enhancer-activating Akt, preventing its apoptotic cleavage and promoting cell survival. Cell Death Differ 14:368-377.
Thau-Zuchman O, Shohami E, Alexandrovich AG, Leker RR (2010) Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J Cereb Blood Flow Metab 30:1008-1016.
Theus MH, Wei L, Francis K, Yu SP (2006) Critical roles of Src family tyrosine kinases in excitatory neuronal differentiation of cultured embryonic stem cells. Exp Cell Res 312:3096-3107.
van Rooijen E, Voest EE, Logister I, Bussmann J, Korving J, van Eeden FJ, Giles RH, Schulte-Merker S (2010) von Hippel-Lindau tumor suppressor mutants faithfully model pathological hypoxia-driven angiogenesis and vascular retinopathies in zebrafish. Dis Model Mech 3:343-353.
Wang Z, Luo F, Li L, Yang L, Hu D, Ma X, Lu Z, Sun L, Cao Y (2010) STAT3 activation induced by Epstein-Barr virus latent membrane protein1 causes vascular endothelial growth factor expression and cellular invasiveness via JAK3 And ERK signaling. Eur J Cancer 46:2996-3006.
Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D, Cheresh D (2004) Src blockade stabilizes a Flk/ cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest 113:885-894.
Wu H, Jiang H, Lu D, Qu C, Xiong Y, Zhou D, Chopp M, Mahmood A (2011) Induction of angiogenesis and modulation of vascular endothelial growth factor receptor-2 by simvastatin after traumatic brain injury. Neurosurgery 68:1363-1371; discussion 1371.
Xiong Y, Zhang Y, Mahmood A, Meng Y, Qu C, Chopp M (2011) Erythropoietin mediates neurobehavioral recovery and neurovascular remodeling following traumatic brain injury in rats by increasing expression of vascular endothelial growth factor. Transl Stroke Res 2:619-632.
Xu JJ, Chen EY, Lu CL, He C (2009) Recombinant ciliary neurotrophic factor promotes nerve regeneration and induces gene expression in silicon tube-bridged transected sciatic nerves in adult rats. J Clin Neurosci 16:812-817.
Zachary I, Gliki G (2001) Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res 49:568-581.
Zan L, Zhang X, Xi Y, Wu H, Song Y, Teng G, Li H, Qi J, Wang J (2014) Src regulates angiogenic factors and vascular permeability after focal cerebral ischemia-reperfusion. Neuroscience 262:118-128.
Zan L, Wu H, Jiang J, Zhao S, Song Y, Teng G, Li H, Jia Y, Zhou M, Zhang X, Qi J, Wang J (2011) Temporal profile of Src, SSeCKS, and angiogenic factors after focal cerebral ischemia: correlations with angiogenesis and cerebral edema. Neurochem Int 58:872-879.
Zhang L, Deng M, Zhou S (2011) Tetramethylpyrazine inhibits hypoxia-induced pulmonary vascular leakage in rats via the ROS-HIFVEGF pathway. Pharmacology 87:265-273.
Zhao H, Bao XJ, Wang RZ, Li GL, Gao J, Ma SH, Wei JJ, Feng M, Zhao YJ, Ma WB, Yang Y, Li YN, Kong YG (2011) Postacute ischemia vascular endothelial growth factor transfer by transferrin-targeted liposomes attenuates ischemic brain injury after experimental stroke in rats. Hum Gene Ther 22:207-215.
Copyedited by Slone-Murphy J, Raye W, Li CH, Song LP, Zhao M
10.4103/1673-5374.175067
How to cite this article: Guo H, Zhou H, Lu J, Qu Y, Yu D, Tong Y (2016) Vascular endothelial growth factor∶ an attractive target in the treatment of hypoxic/ischemic brain injury. Neural Regen Res 11(1)∶174-179.
Funding: Funding: This study was supported by the National Natural Science Foundation of China, No. 81401238, 81330016, 31171020, 81172174 and 81270724; the grants from Ministry of Education of China, No. 313037, 20110181130002; a grant from State Commission of Science Technology of China, No. 2012BAI04B04; the grants from Science and Technology Bureau of Sichuan Province of China, No. 2012SZ0010, 2014FZ0113, 2014SZ0149; and a grant from Clinical Discipline Program (Neonatology) from the Ministry of Health of China, No. 1311200003303.
*Correspondence to: Yu Tong or Dan Yu, zisu_yu@163.com or yd540@126.com.
杂志排行

中国神经再生研究(英文版)的其它文章
- Angiogenesis in tissue-engineered nerves evaluated objectively using MICROFIL perfusion and micro-CT scanning
- Dexamethasone prevents vascular damage in earlystage non-freezing cold injury of the sciatic nerve
- Cerebrolysin improves sciatic nerve dysfunction in a mouse model of diabetic peripheral neuropathy
- A novel bioactive nerve conduit for the repair of peripheral nerve injury
- Treatment with analgesics after mouse sciatic nerve injury does not alter expression of wound healingassociated genes
- Time representation of mitochondrial morphology and function after acute spinal cord injury