理论研究BBPQ-PC61BM体系的光伏性质
2016-11-22赵蔡斌葛红光靳玲侠王文亮尹世伟
赵蔡斌 葛红光 张 强 靳玲侠 王文亮 尹世伟
(1陕西理工学院化学与环境科学学院,陕西省催化基础与应用重点实验室,陕西汉中723000;2陕西师范大学化学化工学院,陕西省大分子科学重点实验室,西安710062)
理论研究BBPQ-PC61BM体系的光伏性质
赵蔡斌1,*葛红光1,*张强1靳玲侠1王文亮2尹世伟2
(1陕西理工学院化学与环境科学学院,陕西省催化基础与应用重点实验室,陕西汉中723000;2陕西师范大学化学化工学院,陕西省大分子科学重点实验室,西安710062)
探索和制备具有高能量转换效率(PCE)的有机太阳能电池体系是有机电子学的重要领域和研究热点。本文利用量子化学和分子动力学计算结合Marcus-Hush电荷传输模型理论研究了BBPQ-PC61BM(BBPQ:7, 12-二((三异丙基甲硅烷基)乙炔基)苯并(g)吡啶并(2′,3′:5,6)吡嗪并(2,3-b)喹喔啉-2(1H)-酮;PC61BM:(6,6)苯基-C61-丁酸甲酯)体系的光伏性质。结果表明,BBPQ-PC61BM体系具有相当大的开路电压(1.22 V)、高的填充因子(0.90)和高的光电转换效率(9%-10%)。此外,本文研究还发现BBPQ-PC61BM体系拥有中等大小的激子结合能(0.607 eV),但相对较小的激子分离和电荷复合重组能(0.345和0.355 eV)。借助于一个简单的分子复合物模型,本文预测BBPQ-PC61BM体系的激子解离速率常数kdis高达1.775×1013s-1,而预测的电荷复合速率常数krec相当小(<1.0 s-1),这表明在BBPQ-PC61BM相界面上,激子解离效率非常高。总之,理论研究表明,BBPQ-PC61BM是一个非常有前途的有机太阳能电池候选体系,值得实验上做出进一步研究。
BBPQ;PC61BM;理论研究;光伏性质;密度泛函理论
1 Introduction
Organic solar cells(OSCs)have attracted continuous interest in the past several decades due to their numerous advantages compared to traditional silicon-based solar cells,such as lightweight,low cost,adjustable properties,and ease of solvent processing1-4.The power conversion efficiency(PCE)is one of most parameters that character the performance of OSC devices,which is directly related with the open-circuit voltage,Voc,short-circuit current density,Jsc,and fill factor,FF.Previous studies have shown that electron-donating materials in high PCE devices with(6,6)-phenyl-C61-butyric acid methyl ester(PC61BM)as acceptor,should possess(1)the strong optical absorption to harvest more sunlight, (2)high hole mobility to transport holes as efficient as possible, (3)low-lying lowest unoccupied molecular orbital(HOMO)level close to-4.0 eV,and(4)low highest occupied molecular orbital (HOMO)level to obtain large Voc5-7.
Recently,Engelhart et al.8synthesized a series of novel nitrogendoped pentacene derivatives.Interestingly,most of these compounds exhibit the very strong optical response and low LUMO level of~4.0 eV,which makes them seem to be very suitable as an ideal electron donor material.In current work,taking the PC61BM as acceptor and 7,12-bis((triisopropylsilyl)-ethynyl)benzo(g)pyrido (2′,3′:5,6)pyrazino(2,3-b)quinoxalin-2(1H)-one(BBPQ)as donor, we carried out systematic quantum chemistry and molecular dynamics investigations for the photovoltaic properties of BBPQPC61BM system in order to verify our speculation.The main objectives of this work are to explore the feasibility of BBPQPC61BM system as a potential organic solar cell.Calculations show that BBPQ is an excellent electron donor material,and the PCE of BBPQ-PC61BM system can theoretically reach up to 9%or more.
2 Computational methods
As is well-known,the density functional theory(DFT)is an accurate formalism that simulates the molecular structures and electronic properties of organic compounds9-11.However,recent studies show that traditional hybrid density functionals,such as B3LYP,are unsuitable to estimate the excited-state properties for large π-conjugated molecules since their non-Coulomb term of exchange functionals dies off too rapidly12-14.Consequently,the long-range-corrected functional(CAM-B3LYP)15coupled with the 6-311G(d,p)basis set was used to calculate the properties of ground state and excited state in this work,which has been verified to be more reliable and accurate than the other hybrid density functionals16-20.For comparison,some results calculated with the B3LYP/6-311G(d,p)method were also provided.To explore the rational geometry of BBPQ-PC61BM complex,a detailed potentialsurface scan was performed between PC61BM and BBPQ with the CAM-B3LYP-D3(BJ)/6-311G(d,p)scheme.As seen in Fig.S1(in Supporting Information),the BBPQ-PC61BM complex is found to be most stable when the centroids distance of BBPQ and PC61BM is at 0.80 nm,which is in good agreement with the recent study21. Then,in subsequent calculations for the BBPQ-PC61BM complex, the centroid distance of BBPQ and PC61BM is invariably fixed at 0.80 nm.Moreover,the influence of molecular orientation was also considered.As shown in Fig.S2(in Supporting Information), the molecular orientation has a very weak influence on the total energy of BBPQ-PC61BMcomplex.Total density of states(TDOS) and partial density of states(PDOS)were visualized with the Multiwfn 3.37 software package22-24.In addition,the direct-coupling(DC)strategy under one-electron approximation and the PW91PW91/6-31G(d)method25,26was used to estimate the charge transfer integral(VDA)in Marcus-Hush model,which have been illustrated to provide the most accurate VDAvalue at the DFT level27,28.All quantum chemistry calculations were completed with the Gaussian 09 software29.
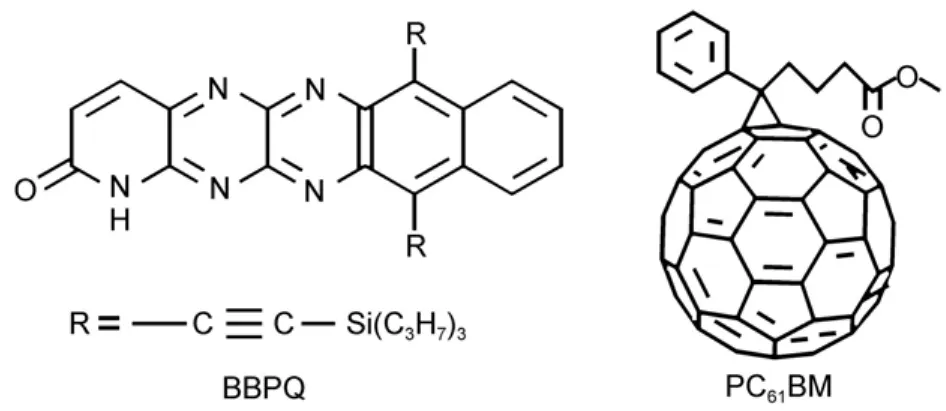
Fig.1 Molecular structures of BBPQ and PC61BM
3 Results and discussion
3.1Electronic properties and open-circuit voltage
The structures of BBPQ and PC61BM were depicted in Fig.1. Our optimization reveals that BBPQ core keeps an excellent planar geometry(Fig.S3,in Supporting Information),which indicates its good electronic delocalization.With the optimized ground-state geometries of PC61BM and BBPQ,the TDOS and PDOS were calculated and presented in Fig.2.With the PDOS,the contribution from each substituent to the frontier molecular orbital can be directly observed.As seen,for PC61BM most density of HOMOs and LUMOs concentrates on the C60spheroid in energyrange from-10.0 to 2.0 eV,and the contribution of substituent (methyl-4-phenylbutanoate)is very small.This result indicates that the substituent only enhances the C60solubility in organic solvents,and has hardly influence on its electronic properties, which is in good accord with the previously experimental result30,31.Furthermore,it is found that,as expected,the contribution to the HOMO and LUMO of BBPQ from the trimethylsilyl is very small,which indicates that the electronic structure of BBPQ is almost completely determined by its core skeleton.According to the previous study,the Vocof OSCs can be estimated with32
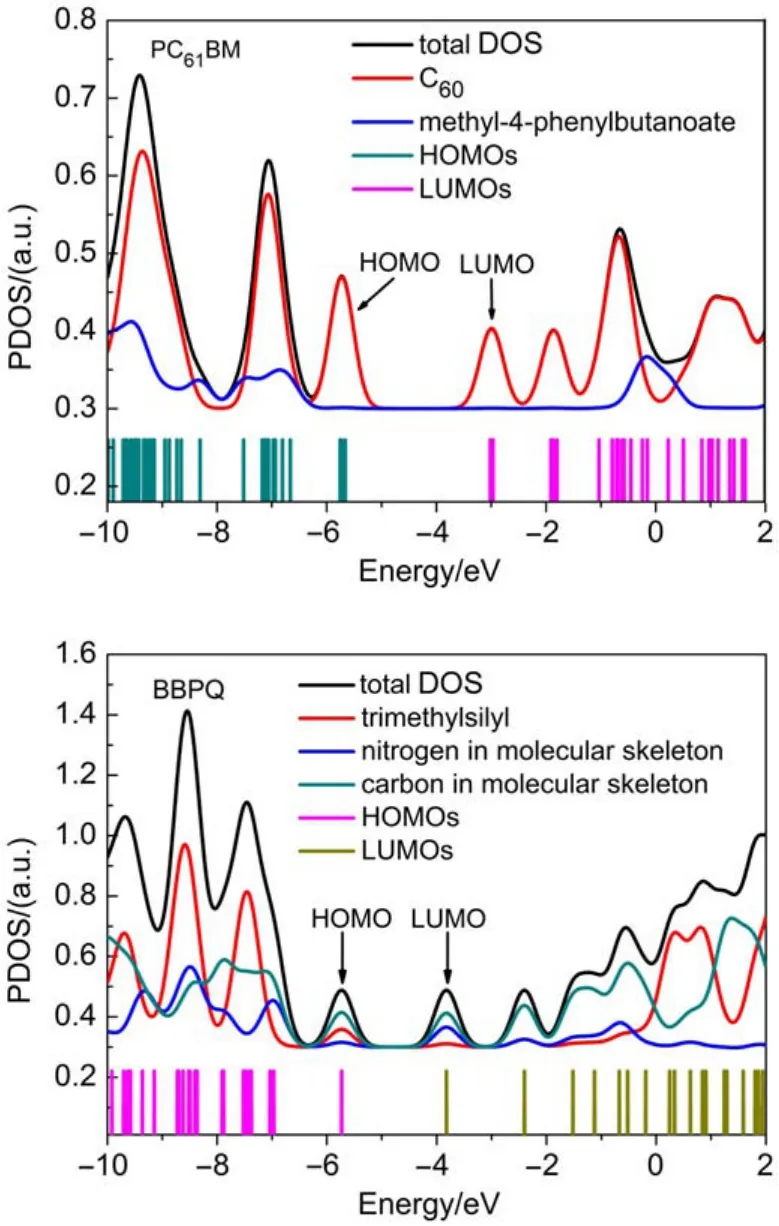
Fig.2 Total and partial density-of-states of PC61BM and BBPQ
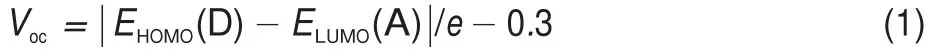
where EHOMO(D)and ELUMO(A)are the HOMO level of donor and the LUMO level of PC61BM,respectively,e is the electronic charge,and the value of 0.3 V is an empirical factor.Then,based on the LUMO level(-4.3 eV)for PC61BM,as well as the HOMO level(-5.82 eV)of BBPQ,the Vocis estimated to be as large as 1.22 V for the BBPQ-PC61BM system.More interestingly,the PCE of BBPQ-PC61BM system is predicted to reach up 9%-10%or more(Fig.3)by means of the Scharber diagram32,which indicates the BBPQ-PC61BM system being a very promising OSC.
3.2Short-circuit current density(JSC)and fill factor(FF)
The Jscis another important factor that determines the PCE of OSC devices.Simply,the Jscis viewed as a function of the lightabsorbing efficiency(η(λ)),internal quantum efficiency(ηIQE(λ)), and the spectral irradiance of incident light(S(λ)),which can be expressed as33-35,
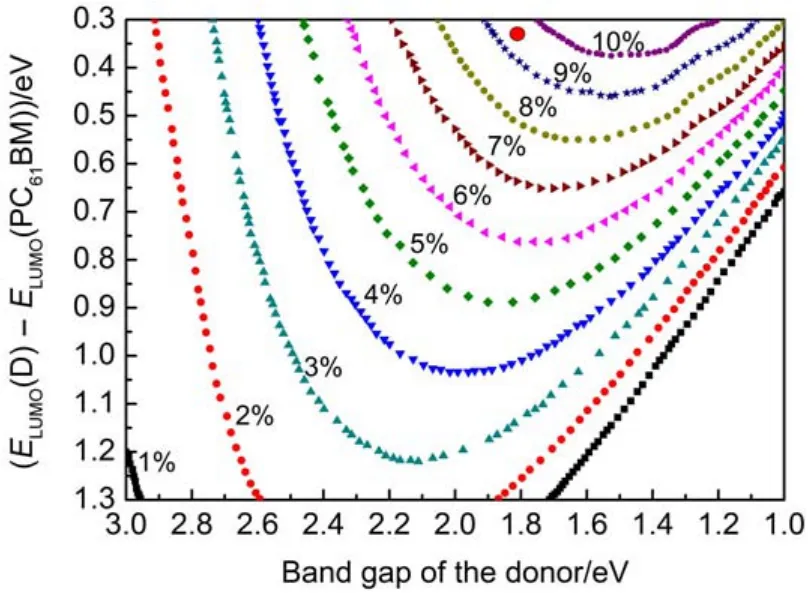
Fig.3 Predicted PCE for BBPQ-PC61BM cell with the Scharber diagram
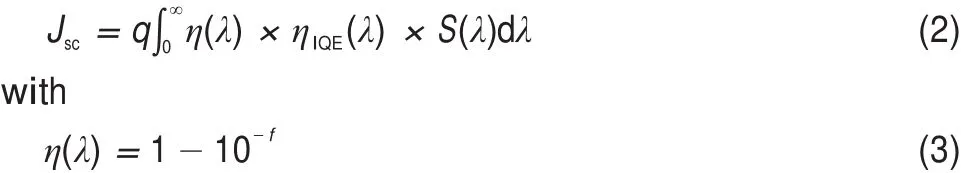
where η(λ)is the light-absorption efficiency,ηIEQ(λ)is the internal quantum efficiency,S(λ)is the spectral irradiance of incident light, and f is the oscillator strength of molecular donor associated with a certain wavelength.From Eq.(2)and Eq.(3),it is clear that the wide and strong optical absorption can remarkably enlarge the Jsc. Here,the η(λ)values of the strongest absorption peak and the second-strongest one were estimated.Calculations show that the η(λ)is equal to 0.55/0.45 for the strongest/second-strongest absorption peak of BBPQ.For the FF calculation,an approximate scheme can be expressed as36,37,

where νocis the dimensionless voltage,which can be estimated by the following equation38,39,

where kB,T,and q are Boltzmann constant,temperature(here,we set T=300 K),and the elementary charge respectively,n is the ideality factor of the diode.According to estimated Voc(1.22 V)for the BBPQ-PC61BM system,the νocis estimated to be 46.42 at n= 1,then,the upper-limit of FF is calculated to be as high as 0.90.
3.3Exciton binding energy
Generally,the exciton dissociation concludes a two-step process,where excitons are firstly separated to less strongly bound polaron pairs and,finally,to free polarons40.In order to dissociate excitons into free polarons,the exciton binding energy(Eb)has to be overcome.In optoelectronic organic devices,the Ebis one of the most important parameters that govern many physical processes,which is directly related to the charge separation efficiency. Usually the exciton binding energy is taken as the difference between the transport gap(Et)and the optical band one(Eopt).The former is the difference between adiabatic ionization potential (EAIP)and adiabatic electron affinity(EAEA),while the latter is taken as the first-singlet excitation energy.According this scheme,the Ebcan be calculated as the following expression41,
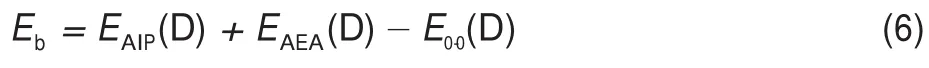
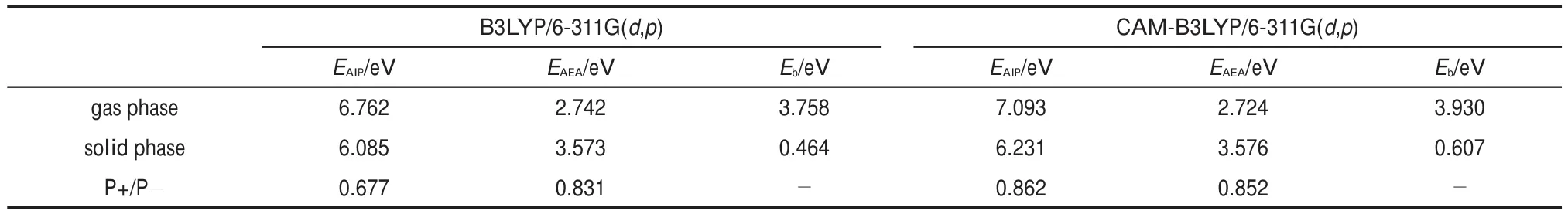
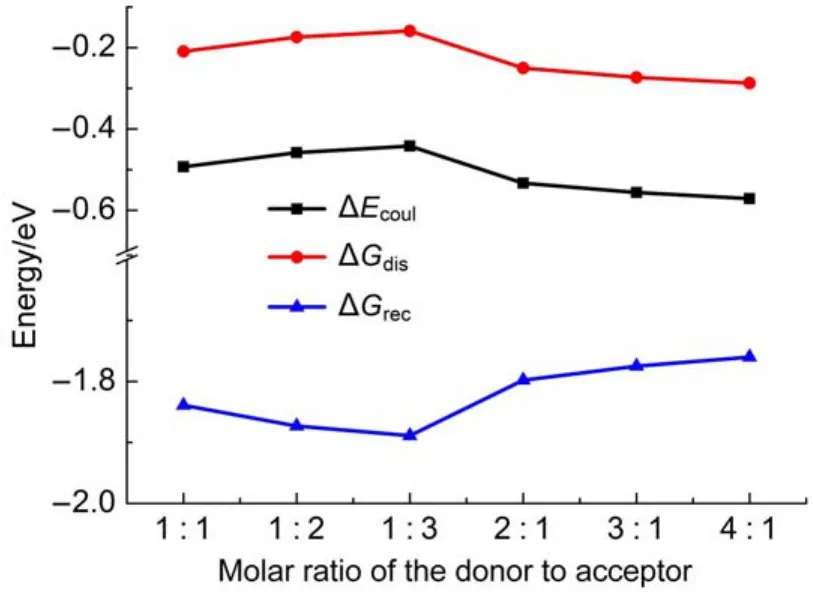
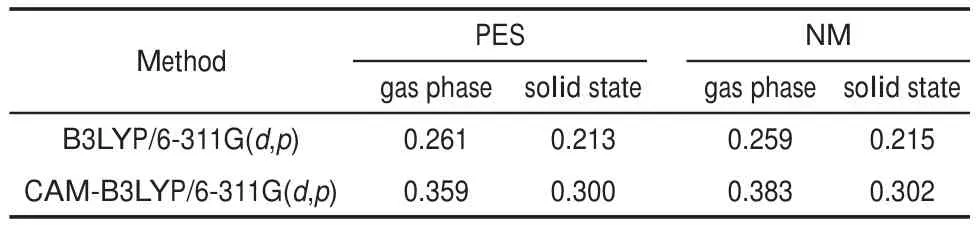
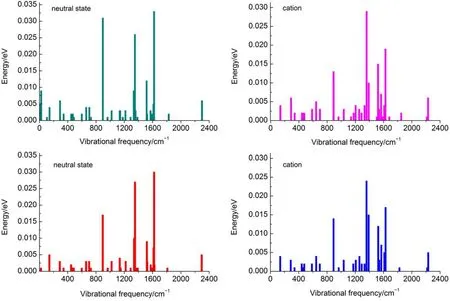
where EAIP(D)and EAEA(D)are the donor′s AIP and AEA in the solid state,and E0-0(D)is the lowest singlet-excited energy of donor.As is well-known,the solid stack can stabilize the ionic species,lower the IP,and increase the EA.Then,to calculate the Eb,the EAIPand the EAEAof solid donor firstly need to estimated. Here,the EAIPand the EAEAof BBPQ in the solid state were calculated via the scheme reported by Schwenn et al.42,which has been verified being an accurate method to estimate the IP and EA of organic materials in the solid state.Calculated EAIPand EAEAvalues as well as the Ebin the solid and gas states for BBPQ with different DFT methods were listed in Table 1.As seen,the EAIP/ EAEAin the gas phase is clearly larger/smaller than the one in the solid state,which indicates relatively large polarization energies (~0.8 eV)from gas phase to solid state.In addition,the estimated Ebis remarkably large regardless of the solid stacking compared to the measured Ebin numerous organic materials43.Thus,it is essential to consider the solid stacking effect for accurately estimating the Eb.The precious study showed that an exciton breaks free the Coulomb attraction and becomes two carriers with an opposite charge when Eb 3.4Gibbs free energies of exciton dissociation and charge recombination The Gibbs free energy change(ΔG)of electron transfer process can be estimated as the energy difference of constituents in the final and initial states,accounting for the Coulombic attraction between the two charges in the charge-separated state.Thus,for the exciton-dissociation process,the ΔG(ΔGdis)is expressed as45, whereED*,ED+,EA,andEA-represent the total energies of the isolated donor in the equilibrium geometries of the lowest singletexcited state and of the cationic state and the total energies of the isolated acceptor in the equilibrium geometries of the ground state and of the anionic states,respectively.qDand qAare the atomic charges(obtained by Mulliken population analysis in this work) on donor and acceptor in their relevant state with a separation rDA, ε0is the vacuum permittivity,and εsis the relative permittivity of material.The ΔG(ΔGrec)in the charge recombination can also be estimated according to the similar expression to Eq.(7)and Eq.(8). For organic compounds,the εscan be accurately estimated by the following Clausius-Mossotti(CM)equation46 where V is the Connolly molecular volume,=13∑αii,αiiis the diagonal matrix element of first-order polarizability tensors. Calculation shows that the εsvalue is 2.451 for the solid BBPQ, which is in good agreement with the measured ones(varying in the range from 2 to 547,48)in most organic photoelectric materials. Since the εsof solid PC61BM cannot be accurately computed with the Eq.(9)due to the so-called“tail effect”,the experimental εsof 3.949is used in current calculation.For the BBPQ-PC61BM complex,the total εsis taken as an average of BBPQ and PC61BM. Fig.4 showed the ΔGdis,ΔGrecas well as the ΔEcoulterm estimated in different BBPQ-PC61BM blends.As seen,in BBPQ-PC61BM complexes the ΔGdisand the ΔGrecvalues are calculated to be consistently negative,which indicates that the exciton-dissociation and charge-recombination processes are always favorable thermodynamically.Furthermore,compared to the ΔGdisandthe ΔGrec, it can be noted that the former is remarkably larger than the latter, which denotes that the driving force of charge-recombination is larger than that of exciton-dissociation for the BBPQ-PC61BM system. Table 1 Calculated EAIPand EAEAvalues as well as the Ebin gas and solid states for BBPQ with different DFT methods 3.5Reorganization energies of exciton dissociation and charge recombination Generally,the total reorganization energy(λ)accompanying the charge transfer in organic materials can be divided into two sections,namely,the internal reorganization energy(λint)and external one(λext).The λintterm can be calculated with the classic adiabatic potential energy surface(PES)method50,51.For example, in the case of exciton dissociation,the λintis actually taken as the average of the following λ1and λ252, where QRand QPrefer to the equilibrium geometries of the reactants(R)and products(P),respectively.Our calculation shows that the λint(λdis)is 0.275 eVin the exciton-dissociation for PC61BMBBPQ complex,which slightly increases to 0.285 eV for the charge recombination.Relatively,the λextis very difficult to be accurately calculated.Here,the λextwas estimated by the classicaldielectric continuum model initially developed by Marcus for electron-transfer reactions between spherical ions in solution. According this model,the λextterm is given by53, Fig.4 ΔGdis,ΔGrec,and ΔEcoulvalues calculated in BBPQ and PC61BM blends with different proportions where εopis the optical dielectric constant of material,RD(=0.62 nm for BBPQ)and RA(=0.65 nm for PC61BM)are the effective radii of donor and acceptor estimated as the radius of the sphere having the same surface as the surface accessible area of molecule. The qDand qAterms denote the atomic charges on the ions.The εopcan be estimated with the Lorentz-Lorenz equation54,55, where n is refractive index,Vmis the molar volume(Vm=M/ρ,M is the molar mass,and ρ is the material density),R is the molar refraction.In this work,the ρ was estimated with the molecular dynamics simulation,and the simulated details were presented in the Supporting Information.Calculations show that the ρ and R of BBPQ solid are 1.066 g·cm-3and 127.4 cm3·mol-1,respectively, yielding the εopof 2.145 for BBPQ.The εopof PC61BM is estimated to be 3.482 with its experimental refractive index of 1.866.Based on the above parameters,the λextis estimated to be 0.060 eV in BBPQ-PC61BM complex(1:1).Summary,the total λ is 0.335 eV in the exciton-dissociation process for BBPQ-PC61BM complex. However,for the charge-recombination process,it further increases to 0.345 eV.According to the Marcus model,the large λ decreases the charge transfer rate;our results show that the exicton-dissociation rate is faster than the charge-recombination one without considering the VDA. 3.6Exciton dissociation and charge recombination rates As is known to all,the charge transfer process occurring in organic solid materials under the high temperature approximation obeys the incoherent hopping mechanism56,57,and the rate constant, k,can be evaluated using the classical Marcus-Hush model58,59, where λ is the total reorganization energy,VDAis the effective charge transfer integration between donor and acceptor,ΔG is the Gibbs free energy difference between the initial and final states, kBis Boltzmann constant,h is Planck constant,and T is the temperature.In the exciton-dissociation and charge-recombination processes,ΔG=ΔGdisand ΔGrec,respectively.In terms of the DC scheme,the VDAin the charge transfer process can be calculated by the following expression60, where TD(i)A(j)is the charge transfer integral of the ith molecular orbital of donor and the jth molecular orbital of acceptor,SD(i)A(j)is the spatial overlap integral of the ith molecular orbital of donor and the jth molecular orbital of acceptor,and eD(i)/eA(j)is the site energy.The TD(i)A(j),SD(i)A(j),and eD(i)/eA(j)can be obtained from the TD(i)A(j)=<ψD(i)|FKS|ψA(j)>,SD(i)A(j)=<ψD(i)|ψA(j)>,and eD(i)/eA(j)=<ψD(i)/ψA(j)|FKS|ψD(i)/ψA(j)>.Among them,ψD(i)is the HOMO or LUMO of donor, ψA(j)is LUMO of acceptor,and FKSis the Kohn-Sham matrix of donor-acceptor system.The FKScan be estimated from where S is the intermolecular overlap matrix,C is the molecular orbital coefficient matrix from the isolated monomer,and ε is the orbital energy from one-step diagonalization without iteration. Consideration the LUMO+1 and LUMO+2 in PC61BM are degenerate in energy with its LUMO,the total VDAwere estimated as an average value of three VDAvalues between the LUMO of BBPQ and the LUMO/LUMO+1/LUMO+2 of PC61BM.Based on the calculated VDAand λ,the exciton-dissociation rate constant, kdis,is estimated to be as high as 1.775×1013s-1in BBPQ-PC61BM blend with a ratio of 1:1,but the charge recombination rate constant,krec,is predicted to be quite small(<1.0 s-1),which indicates very high exciton-dissociation efficiency in BBPQ-PC61BM interface.As observed in Eq.(14),the large kdisvalue can be attributed the large ΔGdis.According our calculations,the excitondissociation process,really occurs in the normal region of Marcus since|ΔG|<λ(0.245 eV versus 0.335 eV).As a result,the k will increase significantly if the|ΔG|and the λ are to converge toward a similar value.Unlike the exciton dissociation,the charge recombination process happens in the inverted region of Marcus due to the|ΔG|>>λ(1.803 eV versus 0.345 eV).Thus,the large|ΔG| remarkably decreases the krec. Table 2 Calculated λintfor BBPQ in solid and gas states with different DFT methods 3.7Charge transport in BBPQ solid As is well known,the charge transport ability of donor also affects remarkably the solar cell performance.According to the previous investigation,for high-performance OSC devices,the hole carrier mobility should be as high as 10-3cm2·V-1·s-1at least32.Hence,we estimated the charge-transport performance by means of calculating the λ and VDAvalues with a simplified dimer model,which has been widely applied to evaluate the charge-transport performance of organic material61-63.Table 2 displayed the calculated λintvalues with the PES and normal mode(NM) analysis.As seen,the λintestimated with two approaches are quite close,which shows that the harmonic oscillator approximation can describe well for the charge transfer process of studied molecule64. In addition,it can be also noticed that the λintin the solid state are obviously smaller than that in the gas state,which indicates that the solid stack can limit the structural relaxation of BBPQ in charge transfer process to a certain extent.Considering the practical operating condition of OSC devices,the λintestimated in the solid state is more reasonable. Fig.5 Contribution of each vibration mode to the λintfor BBPQ calculated in gas(up)and solid(down)states To clarify the λintorigin,the contribution from each vibrational mode to the λintwas calculated with the DUSHIN program developed by Reimers et al.65,66.Fig.5 visualized the contribution from each vibrational mode to the λintestimated at the CAMB3LYP/6-311G(d,p)level in the solid and gas states.As seen, although numerous modes couple with the hole transport in BBPQ,the main contribution to the λintderives from the highfrequency region of 1200-1600 cm-1,which belongs to the stretching vibration of the C―C/C―N single and double bonds located in the molecular skeleton67.Relatively,the contribution from the middle-and low-frequency region is small.Interestingly, from gas state to solid phase,the contribution from the C―C stretching mode with the frequency of 896 cm-1is found to remarkably decrease(from 31 to 16 meV).In addition,the VDAis estimated to be 3.06 meV by means of the face-to-face dimer with the centroids distance of 0.65 nm(Fig.S4 and Fig.S5,in Supporting Information),and then yielding the hole mobility is as high as 1.180×10-3cm2·V·s-1according to the one-dimensional(1D) charge transfer model. In summary,BBPQ-PC61BM as a promising OSC was investigated theoretically by means of quantum-chemical calculations. Results show that BBPQ-PC61BM system possesses a large opencircuit voltage(1.22 V),high fill factor(0.90),and high PCE (>9%).Also it has a middle-sized exciton binding energy(0.607 eV),relatively large Gibbs free-energy difference(-0.245 eV)in the exciton dissociation,but the very small one(-1.803 eV)in the charge recombination.Using the Marcus′s charge transfer model, the exciton-dissociation rate constant,kdis,is predicted to be as large as 1.775×1013s-1in BBPQ-PC61BM interface.However,the charge-recombination one,kdis,is estimated to be very small(<1.0 s-1)under the same condition.Furthermore,by means of the 1D model,the mobility of BBPQ solid is predicted to be as high as 1.180×10-3cm2·V·s-1,which can be attributed its small inner organization energy(0.261 eV)and relatively large VDA(3.06 meV).In a word,our calculation shows that BBPQ-PC61BM is a promising OSC system,and is worth studying further on the experimental aspect. Supporting Information:available free of charge via the internet at http://www.whxb.pku.edu.cn. References (1) Boudreault,P.L.T.;Najari,A.;Leclerc,M.Chem.Mater.2011, 23,456.doi:10.1021/cm1021855 (2)Cheng,Y.J.;Yang,S.H.;Hsu,C.S.Chem.Rev.2009,109, 5868.doi:10.1021/cr900182s (3) Günes,S.;Neugebauer,H.;Sariciftci,N.S.Chem.Rev.2007, 107,1324.doi:10.1021/cr050149z (4) Thompson,B.C.;Fréchet,J.M.J.Angew.Chem.Int.Ed.2007, 47,58.doi:10.1002/anie.200702506 (5) Peet,J.;Senatore,M.L.;Heeger,A.J.;Bazan,G.C.Adv.Mater. 2009,21,1521.doi:10.1002/adma.200802559 (6) Huo,L.;Hou,J.;Chen,H.Y.;Zhang,S.;Jiang,Y.;Chen,T.; Yang,Y.Macromolecules 2009,42,6564.doi:10.1021/ ma9012972 (7) Sista,P.;Nguyen,H,;Murphy,J.W.;Hao,J.;Dei,D.K.; Palaniappan,K.;Servello,J.;Kularatne,R.S.;Gnade,B.E.; Xue,B.F.;Dastoor,P.C.;Biewer,M.C.;Stefan,M.C. Macromolecules 2010,43,8063.doi:10.1021/ma101709h (8) Engelhart,J.U.;Lindner,B.D.;Tverskoy,O.;Rominger,F.; Bunz,U.H.F.Org.Lett.2012,14,1008.doi:10.1021/ ol203334u (9) Fabiano,E.;Sala,F.D.;Cingoland,R.;Weimer,M.;Görling,A. J.Phys.Chem.A 2005,109,3078.doi:10.1021/jp044974f (10) Tsai,F.C.;Chang,C.C.;Liu,C.L.;Chen,W.C.;Jenekhe,S.A. Macromolecules 2005,38,1958.doi:10.1021/ma048112o (11) Hutchison,G.R.;Ratner,M.A.;Marks,T.J.J.Am.Chem.Soc. 2005,127,2339.doi:10.1021/ja0461421 (12)Wong,B.M.;Hsieh,T.H.J.Chem.Theory.Comput.2010,6, 3704.doi:10.1021/ct100529s (13) Grimme,S.;Parac,M.ChemPhysChem 2003,4,292. doi:10.1002/cphc.200390047 (14) Song,J.W.;Hirao,K.Theor.Chem.Acc.2014,133,1438. doi:10.1007/s00214-013-1438-5 (16) Vlček,A.;Záliš,S.Coordin.Chem.Rev.2007,251,258. doi:10.1016/j.ccr.2006.05.021 (17) Zhang,S.;Qu,Z.;Tao,P.;Brooks,B.;Shao,Y.;Chen,X.;Liu, C.J.Phys.Chem.C 2012,116,12434.doi:10.1021/jp3027447 (18) Jacquemin,D.;Perpète,E.A.;Vydrov,O.A.;Scuseria,G.E.; Carlo,A.J.J.Chem.Phys.2007,127,094102.doi:10.1063/ 1.2770700 (19) Jacquemin,D.;Planchat,A.;Adamo,C.;Mennucci,B.J.Chem. Theory.Comput.2012,8,2359.doi:10.1021/ct300326f (20) Jorge,F.E.;Jorge,S.S.;Suave,R.N.Chirality 2015,27,23. doi:10.1002/chir.22384 (21) Liu,T.;Troisi,A.J.Phys.Chem.C 2011,115,2406. doi:10.1021/jp109130y (23) Lu,T.;Chen,F.W.J.Mol.Graph.Model.2012,38,314. doi:10.1016/j.jmgm.2012.07.004 (24) Lu,T.;Chen,F.W.Acta Chim.Sin.2011,69,2393.[卢天,陈飞武.化学学报,2011,69,2393.] (25) Troisi,A.;Orlandi,G.J.Phys.Chem.A 2006,110,4065. doi:10.1021/jp055432g (26)Yin,S.W.;Yi,Y.P.;Li,Q.X.;Yu,G.;Liu,Y.Q.;Shuai,Z.G. J.Phys.Chem.A 2006,110,7138.doi:10.1021/jp057291o (27) Song,Y.B.;Di,C.A.;Yang,X.D.;Li,S.P.;Xu,W.;Liu,Y.Q.; Yang,L.M.;Shuai,Z.G.;Zhang,D.Q.;Zhu,D.B.J.Am. Chem.Soc.2006,128,15940.doi:10.1021/ja064726s (28) Huang,J.S.;Kertesz,M.Chem.Phys.Lett.2004,390,110. doi:10.1016/j.cplett.2004.03.141 (29) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09, Revision D.02;Gaussian Inc.:Wallingford,CT,2009. (30) Zheng,L.P.;Zhou,Q.M.;Deng,X.Y.;Yuan,M.;Yu,G.;Cao, Y.J.Phys.Chem.B 2004,108,11921.doi:10.1021/jp048890i (31)Wang,X.M.;Guo,Y.L.;Xiao,Y.;Zhang,L.;Yu,G.;Liu,Y.Q. J.Mater.Chem.2009,19,3258.doi:10.1039/B823336E (32) Scharber,M.C.;Mühlbacher,D.;Koppe,M.;Denk,P.; Waldauf,C.;Heeger,A.J.;Brabec,C.J.Adv.Mater.2006,18, 789.doi:10.1002/adma.200501717 (33) Peumans,P.;Yakimov,A.;Forrest,S.R.J.Appl.Phys.2003,93, 3693.doi:10.1063/1.1646446 (34) Bérubé,N.;Gosselin,V.;Gaudreau,J.;Côté,M.J.Phys.Chem. C 2013,117,7964.doi:10.1021/jp309800f (35) Liu,X.R.;Shen,W.;He,R.X.;Luo,Y.F.;Li,M.J.Phys. Chem.C 2014,118,17266.doi:10.1021/jp503248a (36) Guo,X.G.;Zhou,N.J.;Lou,S.J.;Smith,J.;Tice,D.B.; Hennek,J.W.;Ortiz,R.P.;Navarrete,J.T.L.;Li,S.Y.; Strzalka,J.;Chen,L.X.;Chang,R.P.H.;Facchetti,A.;Marks, T.J.Nat.Photonics 2013,7,825.doi:10.1038/ nphoton.2013.207 (37) Gupta,D.;Mukhopadhyay,S.;Narayan,K.Sol.Energy Mater. Sol.Cells 2010,94,1309.doi:10.1016/j.solmat.2008.06.001 (38) Zhou,Y.H.;Fuentes-Hernandez,C.;Shim,J.W.;Khan,T.M.; Kippelen,B.Energy Environ.Sci.2012,5,9827.doi:10.1039/ C6EE01428C (39) Liu,X.R.;Huang,C.Z.;Shen,W.;He,R.X.;Li,M.J.Mol. Model.2016,22,15.doi:10.1007/s00894-015-2885-9 (40) Grage,M.M.L.;Zaushitsyn,Y.;Yartsev,A.;Chachisvilis, Sundström,M.V.;Pullerits,T.Phys.Rev.B 2003,67,205207. doi:10.1103/PhysRevB.67.205207 (41) Nayak,P.K.;Periasamy,N.Org.Electron.2009,10,1396. doi:10.1016/j.orgel.2009.06.011 (42) Schwenn,P.E.;Burn,P.L.;Powell,B.J.Org.Electron.2011, 12,394.doi:10.1016/j.orgel.2010.11.025 (43) Hill,I.G.;Kahn,A.;Soos,Z.G.;Pascal,R.A.Chem.Phys. Lett.2000,327,181.doi:10.1016/S0009-2614(00)00882-4 (44) Li,Y.Z.;Pullerits,T.;Zhao,M.Y.;Sun,M.T.J.Phys.Chem.C 2011,115,21865.doi:10.1021/jp2040696 (45) Lemaur,V.;Steel,M.;Beljonne,D.;Brédas,J.L.;Cornil,J.J.Am.Chem.Soc.2005,127,6077.doi:10.1021/ja042390l (46) Rysselberghe,P.V.J.Phys.Chem.1931,36,1152.doi:10.1021/ j150334a007 (47) Zang,D.Y.;So,F.F.;Forrest,S.R.Appl.Phys.Lett.1991,59, 823.doi:10.1063/1.105274 (48) Brocks,G.;van den Brink,J.;Morpurgo,A.F.Phys.Rev.Lett. 2004,93,146405.doi:10.1103/PhysRevLett.93.146405 (49) Mihailetchi,V.;van Duren,J.;Blom,P.;Hummelen,J.;Janssen, R.;Kroon,J.;Rispens,M.;Verhees,W.;Wienk,M.Adv.Funct. Mater.2003,13,43.doi:10.1002/adfm.200390004 (50) Malagoli,M.;Brédas,J.L.Chem.Phys.Lett.2000,327,13. doi:10.1016/S0009-2614(00)00757-0 (51) Lemaur,V.;da Silva Filho,D.A.;Coropceanu,V.;Lehmann, M.;Geerts,Y.;Piris,J.;Debije,M.G.;van de Craats,A.M.; Senthilkumar,K.;Siebbeles,L.D.A.;Warman,J.M.;Brédas,J. L.;Cornil,J.J.Am.Chem.Soc.2004,126,3271.doi:10.1021/ ja0390956 (52) Brédas,J.L.;Beljonne,D.;Coropceanu,V.;Cornil,J.Chem. Rev.2004,104,4971.doi:10.1021/cr040084k (56) Tauber,M.J.;Kelley,R.F.;Giaimo,J.M.;Rybtchinski,B.; Wasielewski,M.R.J.Am.Chem.Soc.2006,128,1782. doi:10.1021/ja057031k (57) Coropceanu,V.;Cornil,J.;da Silva Filho,D.A.;Olivier,Y.; Silbey,R.;Brédas,J.L.Chem.Rev.2007,107,926. doi:10.1021/cr050140x (60) Yin,S.W.;Li,L.L.;Yang,Y.M.;Reimers,J.R.J.Phys.Chem. C 2012,116,14826.doi:10.1021/jp303724r (61) Olivier,Y.;Lemaur,V.;Brédas,J.L.;Cornil,J.J.Phys.Chem.A 2006,110,6356.doi:10.1021/jp0571933 (62) Liu,H.G.;Kang,S.;Lee,J.Y.J.Phys.Chem.B 2011,115, 5113.doi:10.1021/jp1045595 (63) Chen,X.K.;Zou,L.Y.;Ren,A.M.;Fan,J.X.Phys.Chem. Chem.Phys.2011,13,19490.doi:10.1039/C1CP22227A (64) Li,H.X.;Zheng,R.H.;Shi,Q.J.Phys.Chem.C 2012,116, 11886.doi:10.1021/jp301536z (65) Weber,P.;Reimers,J.R.J.Phys.Chem.A 1999,103,9830. doi:10.1021/jp991404k (66) Cai,Z.L.;Reimers,J.R.J.Phys.Chem.A 2000,104,8389. doi:10.1021/jp000962s (67)Yang,X.D.;Wang,L.J.;Wang,C.L.;Long,W.;Shuai,Z.G. Chem.Mater.2008,20,3205.doi:10.1021/cm8002172 Theoretical Investigation on Photovoltaic Properties of the BBPQ-PC61BM System ZHAO Cai-Bin1,*GE Hong-Guang1,*ZHANG Qiang1JIN Ling-Xia1WANG Wen-Liang2YIN Shi-Wei2 Exploring and fabricating organic solar cell devices with the high power conversion efficiency(PCE) has kept a major challenge and hot topic in organic electronics research.In this study,we have used quantum chemical and molecular dynamics calculations in conjunction with the Marcus-Hush charge transfer model to investigate the photovoltaic properties of BBPQ-PC61BM.The results revealed that the BBPQ-PC61BM(BBPQ: 7,12-bis((triisopropylsilyl)-ethynyl)benzo(g)pyrido(2′,3′:5,6)pyrazino(2,3-b)quinoxalin-2(1H)-one;PC61BM:(6, 6)-phenyl-C61-butyric acid methyl ester)system theoretically possesses a large open-circuit voltage(1.22 V), high fill factor(0.90),and high PCE of 9%-10%.The calculations also reveal that the BBPQ-PC61BM system has a medium-sized exciton binding energy(0.607 eV),with relatively small reorganization energies(0.345 and0.355 eV)for its exciton-dissociation and charge-recombination processes.Based on a simplified molecular complex,the exciton dissociation rate constant,kdis,was estimated to be as large as 1.775×1013s-1at the BBPQPC61BM interface.In contrast,the charge-recombination rate constant,krec,was very small under the same conditions(<1.0 s-1),which indicated a rapid and efficient exciton-dissociation process at the donor-acceptor interface.Overall,our calculations show that the BBPQ-PC61BM system is a very promising organic solar cell system that is worthy of further research. May 13,2016;Revised:July 4,2016;Published online:July 5,2016. s.ZHAO Cai-Bin,Email:zhaocb@snut.edu.cn;Tel:+86-916-2641660.GE Hong-Guang,Emai:gehg@snut.edu.cn; BBPQ;PC61BM;Theoretical investigation;Photovoltaic property;Density functional theory O641 10.3866/PKU.WHXB201607051 Tel:+86-916-2641660. The project was supported by the National Natural Science Foundation of China(21373132,21502109),Doctor Research Start Foundation of Shaanxi University of Technology,China(SLGKYQD2-13,SLGKYQD2-10,SLGQD14-10),and Education Department of Shaanxi Provincial Government Research Projects,China(16JK1142). 国家自然科学基金(21373132,21502109),陕西理工学院博士科研启动基金(SLGKYQD2-13,SLGKYQD2-10,SLGQD14-10)和陕西省教育厅专项科研计划(16JK1142)资助项目©Editorial office ofActa Physico-Chimica Sinica (15) Yanai,T.Chem.Phys.Lett.2004,393,51.10.1016/j. cplett.2004.06.011 (22) Lu,T.;Chen,F.W.J.Comput.Chem.2012,33,580. 10.1002/jcc.22885 (53) Marcus,R.A.J.Chem.Phys.1965,43,679.10.1063/ 1.1696792 (54) Lorentz,H.A.Ann.Phys.1880,9,641.10.1002/ 18802450406 (55) Lorenz,L.Ann.Phys.1880,11,70.10.1002/18802470905 (58) Marcus,R.A.Rev.Mod.Phys.1993,65,599. RevModPhys.65.599 (59) Hush,N.S.J.Chem.Phys.1958,28,962.10.1063/ 1.1744305
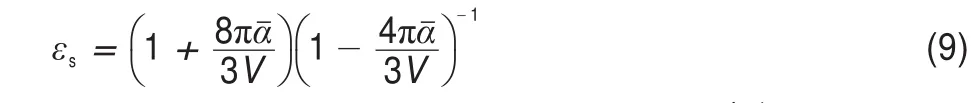
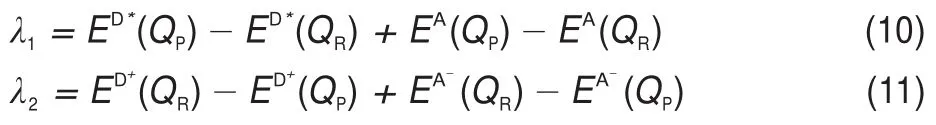
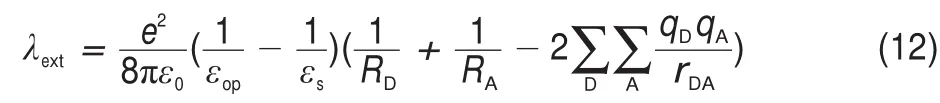

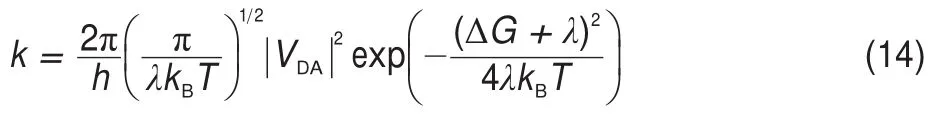
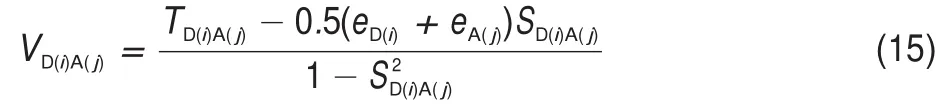

4 Conclusions
(1Shaanxi Province Key Laboratory of Catalytic Fundamentals and Applications,School of Chemical and Environmental Science, Shaanxi University of Technology,Hanzhong 723000,Shaanxi Province,P.R.China;2Key Laboratory for Macromolecular Science of Shaanxi Province,School of Chemistry and Chemical Engineering, Shaanxi Normal University,Xi'an 710062,P.R.China)
