温度对GaN(0001)表面吸附生长TiO2分子的影响*
2016-11-12梁晓琴周金君
梁晓琴,周金君,黄 平,杨 春
(1. 四川师范大学 化学与材料科学学院,成都 610068; 2. 四川师范大学 物理与电子工程学院,成都 610068)
温度对GaN(0001)表面吸附生长TiO2分子的影响*
梁晓琴1,周金君1,黄 平2,杨 春1
(1. 四川师范大学 化学与材料科学学院,成都 610068; 2. 四川师范大学 物理与电子工程学院,成都 610068)
采用基于第一性原理的从头计算分子动力学方法,模拟了在300,400,500,600和700 ℃ 5种温度下TiO2分子在GaN(0001)表面吸附的动力学过程,研究了吸附过程中系统能量、动力学轨迹、Mulliken布居分析、表面成键电子密度分布(ELF)以及扩散系数等性质。结果表明,温度会影响TiO2分子中两个O原子与GaN表面两个Ga原子成键顺序;500 ℃时O2—Ga3成键的时间最早,TiO2在GaN(0001)表面吸附生长的速率最快。600 ℃下TiO2分子在物理吸附阶段的扩散系数比在500 ℃下扩大了接近100倍,500 ℃时O—Ga1的局域电荷分布ELF值最大为0.750,说明500 ℃下O—Ga1键共价性最强。由此可见,TiO2分子在GaN(0001)吸附生长最佳温度是500 ℃。
TiO2;GaN(0001);温度; 动力学; 吸附
0 引 言
在GaN上生长具有丰富物理性能的氧化物铁电材料已受到广泛关注[1-5],GaN(0001)表面外延生长SrTiO3(STO)和 BaTiO3(BTO)薄膜,插不插入缓冲层TiO2对外延薄膜质量有很重大的影响[5],不插缓冲层时晶格失配度很大,而插入缓冲层后,晶格失配度大幅度降低只有1.3%,缓冲层的插入也对生长所需温度影响很大,可以降低外延所需的温度,大幅度提高在GaN表面外延生长铁电薄膜的质量。TiO2在衬底GaN表面的运动轨迹,吸附过程,表面成键电子密度分布和扩散系数等对薄膜的质量有重大影响,而通过薄膜材料的实验还没有办法直接得到吸附粒子微观的物理化学性质,所以采用基于第一性原理的动力学计算,在原子尺度上获得粒子在生长初期物理和化学吸附过程,对薄膜生长的界面控制有着重要的意义[4,6]。
吸附粒子在GaN表面吸附和扩散所消耗的主要能量由基片温度提供,温度对薄膜制备过程中小分子迁移速率、扩散速率和吸附结晶状况3个方面有显著的影响,同时也对集成薄膜的质量有较大的影响,制备薄膜需要合适的基底温度[7]。 在李言荣[8]的实验中,直接在GaN表面外延生长STO薄膜,采用RHEED对STO薄膜的生长情况进行原位监控,发现当温度从700 ℃降低到600 ℃时,STO薄膜衍射强度明显变弱,降低至500 ℃时,STO薄膜在GaN表面吸附生长的过程中都未出现明显的衍射图案,说明500 ℃是STO在GaN表面形成的是非晶膜,GaN基底的温度太高,在直接生长STO薄膜时,极易发生明显的界面扩散现象。当以TiO2为缓冲层制备STO薄膜时,发现当温度低至400 ℃时,STO薄膜的衍射图案依然明亮清晰,TiO2缓冲层有效地降低了STO薄膜的外延生长所需的最低温度,有效的遏制了界面的扩散反应,大幅度的提高了STO薄膜的质量。
温度越高,吸附粒子的能量越大,越容易生成晶化的外延薄膜,薄膜缺陷越少,当温度低于一定程度,多生成的非晶结构的薄膜,对薄膜的择优取向生长不利。如何选择合适的衬底温度来提高薄膜制备的质量?本文采用动力学DFT方法,探讨在300,400,500,600和700 ℃等不同温度TiO2在GaN(0001)表面的吸附过程,计算分析了小分子的吸附轨迹、吸附体系能量的变化、Mulliken布局分析、电子局域分布ELF和不同粒子的扩散系数。初步得出温度对小分子对GaN(0001)体系吸附过程的影响,为实验的进行和改进提供了强有力的理论支持。
1 物理模型和计算方法
1.1 物理模型
本文所采用的GaN(0001)基底是在量子化学薄膜材料计算中普遍应用的薄膜表面,晶格参数a=b=0.321 nm,c=0.523 nm,以Ga原子为终止原子的GaN(0001)表面。固定GaN(0001)基底最下面6层,而充分弛豫接近基底表面的2层GaN分子和TiO2分子。为了准确观测TiO2分子在GaN(0001)表面的生长吸附动力学过程,将键长0.1668 nm,键角为110.08°的气态TiO2分子,平置放入基片上方,与基片表面相距0.30 nm,设真空层高度为3.0 nm,计算的模型如图1所示,将TiO2分子水平放置在GaN表面正上方,Ti原子位于基片表面的中心fcc位上方,两个O原子位于基片表面两个Ga原子上方。
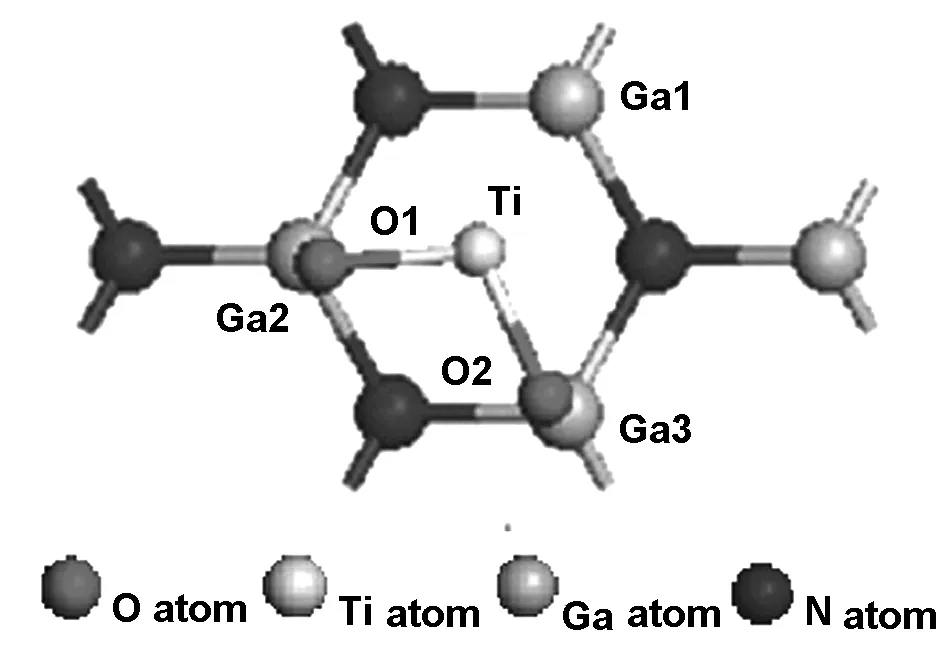
图1 TiO2分子在GaN(0001)表面初始吸附模型的俯视图
1.2 计算方法
本文采用Material Studio中的CASTEP[9-10]软件包进行计算。该计算基于密度泛函理论的平面波超软赝势方法,即用超软赝势[11]描述粒子实与价电子之间的相互作用。电子交换的相关项选择广义梯度近似修正方法为PW91[12-13]形式。收敛精度设为2.0×10-6eV/atom, 平面波截止能量Ecut设置为300 eV。布里渊区k-point 取值为4×4×1,即16个k-point,系综选择NVT,动力学计算步长设为1.0 fs,模拟总时间为1.5 ps,总步数为1 500步。
2 结果与讨论
2.1 温度对吸附动力学过程的影响
用从头计算动力学方法模拟了5个温度下TiO2在GaN基底表面的吸附过程。下面以500 ℃为例,体系能量变化如图2,各个吸附阶段的状态轨迹如图3所示。由图可见整个吸附过程可分为4个阶段:(1) 在0~0.044 ps为物理吸附过程,TiO2分子被GaN表面上的Ga原子和N原子所吸引,逐渐向衬底靠近。TiO2分子中的O原子逐渐向基底表面靠近,其位置比吸附前下降了约0.086 nm,两个Ti—O键的键长由0.1668 nm分别增加到0.1704和0.1751 nm,表明Ti—O键的强度被减弱,主要是由GaN表层的Ga3+与TiO2分子中Ti4+,O2-的静电作用;(2) 在0.044~0.093 ps为O2原子与Ga3原子的成键过程,TiO2分子中的两个O原子继续向下移动,Ti原子偏离吸附前的位置向中心靠近,在0.045 ps时O2—Ga3键开始成键,键长为0.2120 nm,比Ga2O3体材料中Ga—O(0.18~0.21 nm)键长略大,表明O2—Ga3键已经开始部分成键。在0.053 ps时,O2—Ga3键长为0.1920 nm,此时O2—Ga3已经形成较稳定的化学键;(3) 在0.094~0.220 ps时为O1—Ga2键的成键过程,O1原子继续缓慢向下移动,在0.095 ps时,O1—Ga2键开始成键,键长为0.1840 nm,表明O1—Ga2已经形成较稳定的化学键;(4) 在0.220 ps以后,开始形成稳定的吸附态,TiO2分子中的Ti原子最终的平衡位置是GaN表面的中心,TiO2分子中两个O原子分别先后与基底表面的两个Ga原子成键,在0.811 ps,O1—Ga2与O2—Ga3键长最短,系统的能量最低为-39 692.90254 eV。

图2 500 ℃下TiO2在GaN表面的吸附过程与体系能量变化图

图3 500 ℃下TiO2分子吸附过程4个阶段的状态轨迹图
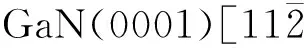
在不同温度下,O1—Ga2与O2—Ga3两个键成键的先后顺序不同,在300 和700 ℃时TiO2分子中的O1先与基底表面的Ga2成键,O2后与基底表面的Ga3成键,在400,500和600 ℃时,顺序恰好相反,说明温度对TiO2在GaN(0001)表面的吸附过程有影响。温度也对O—Ga原子成键的时间有影响,300,400,500,600和700 ℃时,O—Ga开始成键的时间分别为0.052~0.103,0.082~0.143,0.044~0.093,0.048~0.108和0.059~0.126 ps,说明500 ℃时O2—Ga3成键的时间最早,该温度下TiO2在GaN(0001)表面吸附生长的速率最快。
2.2 温度对吸附能的影响
吸附能可以根据下列公式计算得到
在上述公式中,Eadsorbate是吸附前吸附物质TiO2分子的能量,Esurface是GaN(0001)吸附前基片的能量,Eadsorption system是吸附后体系的能量。

表1 不同温度下的吸附能
不同温度下TiO2在GaN(0001)表面的吸附能在7.8~7.9 eV之间,差异不大,说明TiO2能在GaN(0001)表面发生稳定的化学吸附。且TiO2在GaN(0001)表面的吸附能明显大于SrO和BaO分子在GaN(0001)表面的吸附能, 说明TiO2分子在GaN(0001)基片表面优先吸附,这与其它文献的理论计算相符[14]。
2.3 温度对表面电荷与成键的影响
表2为TiO2分子与GaN(0001)表面Ga原子吸附前后Mulliken电荷。从表2可以看出,500 ℃时O1和O2原子的电荷由吸附前的6.56 a.u.分别增加到6.68和6.66 a.u.,增加了0.13和0.11 a.u.,Ti从吸附前的10.82 a.u.增加到11.28 a.u.。数据分析显示在所有温度下,吸附后TiO2分子中的O1、O2、Ti的Mulliken电荷均增大,但不同温度下TiO2增幅不同,说明吸附过程中 TiO2分子得到电子。基底表面的Ga原子的Mulliken电荷由吸附前的12.11 a.u.降低到11.93~12.03 a.u.,表明Ga原子失去电子。从电荷分析来看,吸附过程中TiO2分子从GaN(0001)表面的Ga原子得到电子。
表2 TiO2分子与GaN(0001)表面Ga原子吸附前后Mulliken电荷(a.u.)
图4为不同温度下TiO2分子与GaN表面成键电子密度分布(ELF)。从图4可以看出,吸附后各价电子的局域电荷分布,其中ELF值为0.000~1.000,蓝色代表电荷密度较小的区域,红色代表电荷密度较大的区域,在300~700 ℃下,TiO2分子中的O原子与基底表面Ga原子形成的化学键之间的ELF值依次为0.688,0.710,0.750,0.719和0.680,电子云又密集在O和Ga之间,O—Ga键表现出明显的共价键特征,又因为O的电负性与Ga的电负性相差较大,所以Ga与O形成的化学键又有一定的离子键成分,而TiO2吸附成键后,Ti和O之间的电子密度稍微增加,Ti—O键共价作用增强。由此可见,500 ℃时,成键电子密度分布(ELF)为0.750是最大的,最有利于成键。
2.4 温度对扩散的影响
由于小分子TiO2在GaN(0001)基底表面发生强烈的化学吸附,因此TiO2分子在GaN(0001)表面发生长时间停留,并且TiO2分子不断地在基底表面扩散,这个过程是一个热激活扩散过程,TiO2分子和基底GaN(0001)的扩散能力主要通过扩散系数显示,扩散系数越大,扩散能力也越强。本文采用第一性原理分子动力学方法模拟了在不同温度(300~700 ℃)下TiO2分子中Ti和O原子的吸附过程和移动轨迹,得出5种温度下TiO2中Ti原子和O原子在物理吸附阶段和化学吸附阶段的扩散距离和平均扩散激活能。扩散系数[15]
式中,D0为扩散因子,a为原子移动距离,ΔE为平均扩散激活能,kB为波尔兹曼常数,T为温度,h为普朗克常数,根据此公式能够计算出TiO2分子中Ti和O原子的扩散系数,带*号的为吸附后的相应数据(见表3所示)。
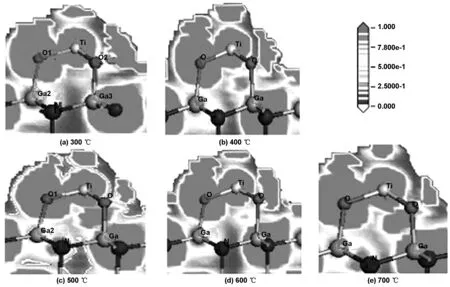
图4 不同温度下TiO2分子与GaN表面成键(O—Ga)电子密度分布(ELF)
500 ℃时在物理吸附阶段TiO2分子中Ti原子与O原子的扩散系数分别为1.03×10-17m2/s和1.65×10-17(1.53×10-17)m2/s。所有温度下的数据分析显示物理吸附阶段TiO2分子中O原子的扩散系数远大于Ti原子的,O1的扩散系数又明显大于O2的扩散系数;在低温阶段(300~400 ℃)TiO2分子中各原子的扩散系数都很小(10-21~10-20m2/s),随着温度的升高(500~700 ℃)扩散系数明显增大(10-17~10-15m2/s),扩散系数最多相差106倍,可见温度对扩散有着不可忽视的影响。TiO2分子在物理吸附阶段的扩散系数(10-21~10-15m2/s)远大于化学吸附阶段的扩散系数(10-18~10-25m2/s),TiO2分子在GaN(0001)表面的吸附过程是一个扩散系数逐渐减小的过程,整个吸附过程可依次分为物理吸附、化学吸附和稳定吸附态。
500~600 ℃时在物理吸附过程中O1的扩散系数从500 ℃时的1.65×10-17增加到600 ℃时的3.83×10-15,增加了近100倍;Ti原子的扩散系数从500 ℃时的1.03×10-17,600 ℃时增加到2.23×10-17,变化很小。而在化学吸附阶段时Ti原子的扩散系数从500 ℃时的2.51×10-20增加到600 ℃时5.92×10-18,增加了近100倍,O1和O2氧原子的扩散系数从500 ℃时的6.86×10-22和2.77×10-22分别增加到6.69×10-19和4.90×10-21,分别增大了10~100倍。由此可见,在600 ℃时TiO2分子中Ti原子与O原子在物理吸附和化学吸附的扩散系数比在500 ℃下显著增大,说明在600 ℃时,O原子的解吸附作用增大,不利于TiO2分子在GaN表面的吸附。在500 ℃时TiO2的扩散系数较小,同时500 ℃时O—Ga1原子的电子密度ELF值最大为0.750,得出TiO2在GaN表面吸附生长最佳生长温度是500 ℃。而且李言荣[8]在STO薄膜制备过程中,用RHEED监控不同温度下TiO2在GaN表面的吸附情况,发现在500 ℃左右,TiO2的衍射图样最为明亮尖锐,说明TiO2的最佳实验生长温度为500 ℃,这与本文理论计算分析得出500 ℃是最优生长温度吻合。
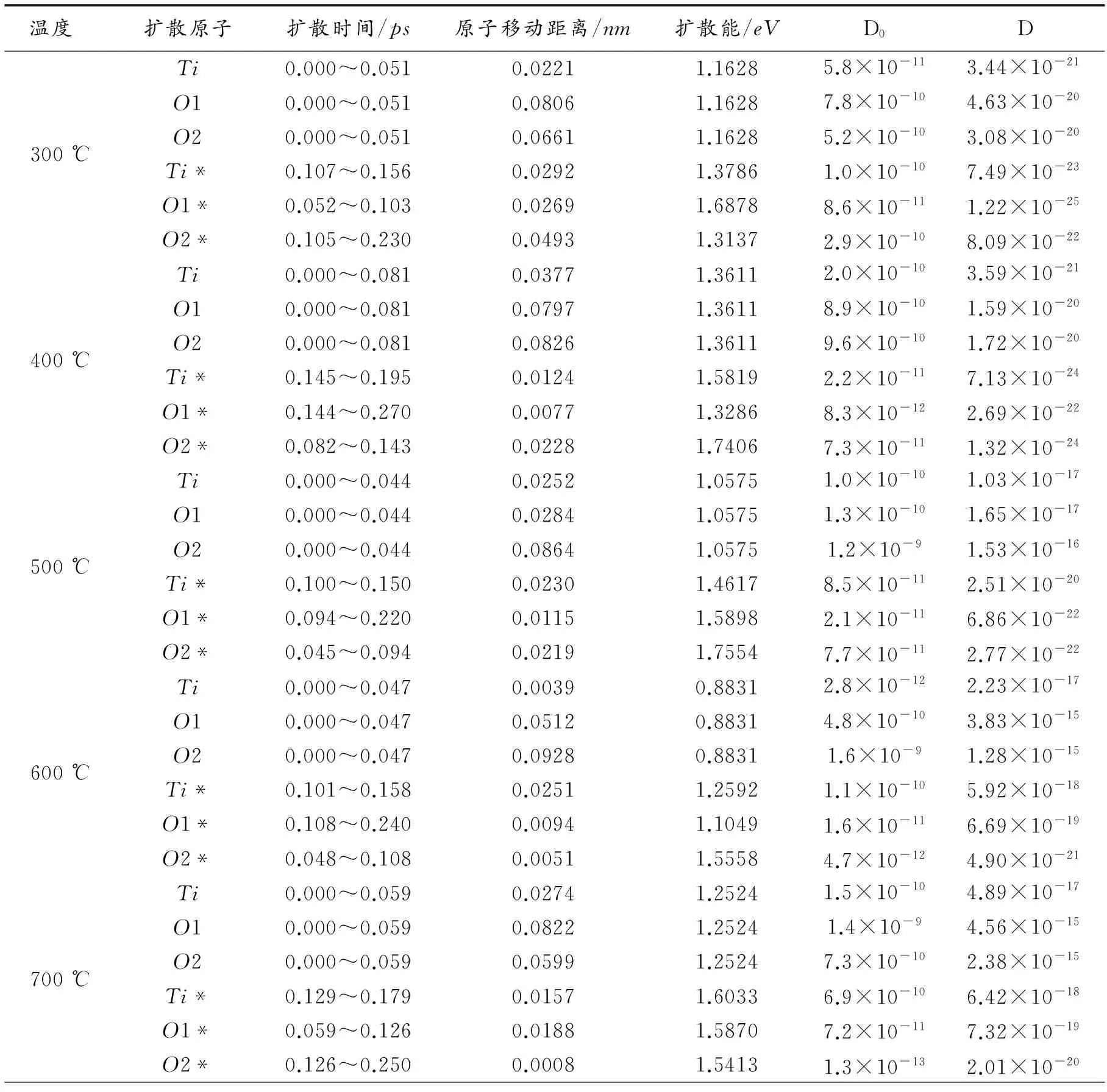
表3 分子在吸附过程中Ti、O的扩散距离和扩散系数
3 结 论
在300~700 ℃下,采用动力学方法模拟了TiO2分子在GaN(0001)表面的吸附过程, 研究了温度对原子运动轨迹、吸附能、界面电荷分布、稳定吸附方位和扩散系数的影响。

[1]YounCJ,JeongTS,HartMS,etal.InfuenceofvariousactivationtemperaturesontheopticaldegradationofMgdopedInGaN/GaNMQWblueLEDs[J].JCrystGrowth,2003,250(3):331-338.
[2]TianW,VaithyanathanV,SchlomDG,etal.Epitaxialintegrationof(0001)BiFeO3with(0001)GaN[J].ApplPhysLett,2007,90(17):172908.
[3]YangSY,ZhanQ,YangPL,etal.Capacitance-voltagecharacteristicsofBiFeO3/SrTiO3/GaNheteroepitaxialstructures[J].ApplPhysLett,2007,91(02):022909.
[4]YangChun,LiYanrong.Computersimulationandthemodelingofthinfilmgrowth[J].JournalFunctionalMaterials,2003,34(3): 247-249.
杨 春,李言荣.薄膜生长模型与计算机模拟[J].功能材料,2003,34(3):247-249.
[5]LuoWB,ZhuJ,ChenH,etal.ImprovedcrystallinepropertiesoflasermolecularbeamepitaxygrownSrTiO2byrutileTiO2layeronhexagonalGaN[J].JApp,2009,106(10):104120-104124.
[6]FengYufang,YangChun,YuYi.DynamicsstudyoftheadsorptionanddiffusioninearlygrowthstageofAlN/α-Al2O3(0001)films[J].ActaPhysSin, 2009,58(5):3553-3559.
冯玉芳, 杨 春, 余 毅.动力学研究AlN/α-Al2O3(0001)薄膜生长初期的吸附与扩散[J].物理学报,2009,58(5): 3553-3559.
[7]WuZiqin,WangBing,SunXia.Filmgrowth[M].Beijing:SciencePress:Beijing, 2013: 190-198.
吴自勤, 王 兵,孙 霞. 薄膜生长[M]. 北京: 科学出版社,2013:190-198.
[8]LiYR,ZhuJ,LuoWB,etal.StudyonthegrowthofintegrateddielectricfilmsonGaNsubstrates[J].RareMetalsLetters,2012,31(2): 45-53.
[9]PayneMC,TeterMP,AllanDC,etal.Iterativeminimizationtechniquesforabinitiototal-energycalculations:moleculardynamicsandconjugategradients[J].RevModPhys,1992, 64(4):1045-1050.
[10]ClarkeLJ,TichI,PayneMC.Large-scaleAbinitiototalenergycalculationsonparallelcomputers[J].ComputPhysCommun, 1992, 72(1): 14-28.
[11]VanderbiltD.Softself-consistentpseudopotentialsinageneralizedeigenvalueformalism[J].PhysRevB,1990,41(11): 7892-7895.
[12]PerdewJP,ChevaryJA,VoskoSH,etal.Atoms,molecules,solids,andsurface:applicationofthegeneralizedgradientapproximationforexchangeandcorrelation[J].PhysRevB, 1992, 46(11): 6671-6687.
[13]PerdewJP,WangY.Accurateandsimpleanalyticrepresentationoftheelectron-gascorrelationenergy[J].PhysRevB, 1992, 45(23): 13244-13249.
[14]HuangP,YangC.TheoreticalresearchofTiO2adsorptiononGaN(0001)surface[J].ActaPhysSin, 2011,60(10): 106801-106806.
黄 平,杨 春.TiO2分子在GaN(0001)表面吸附的理论研究[J].物理学报,2011,60(10): 106801-106806.
[15]ErichW,WalterW,JürgenS.Temperature-dependentdiffusioncoefficientsfromabinitiocomputations:hydrogen,deuterium,andtritiuminnickel[J].PhysReVB,2008, 77:134305-134316.
Effect of temperature on the adsorption of TiO2molecule on GaN(0001) surface
LIANG Xiaoqin1,ZHOU Jinjun1,HUANG Ping2,YANG Chun1
(1. College of Chemistry and Material Science, Sichuan Normal University, Chengdu 610068, China;2. College of Physics and Electronic Engineering, Sichuan Normal University, Chengdu 610068, China)
The dynamics simulates of adsorption process of TiO2molecule on GaN(0001) surfaces in 300, 400, 500, 600 and 700 ℃ temperature are studied by Ab initio molecular dynamics methods based on the first priciples. The energies of system, dynamic trajectories, the analysis of Mulliken populations, the surface bonding electron density distributions (ELF) and diffusion coefficients in the adsorption process are calculated and discussed. The results show that the bonding sequence of O and Ga atoms are different in the different temperature. The time of O2-Ga3 formation is the earliest and the rate of TiO2adsorption on GaN (0001) surface is the highest in 500 ℃. The diffusion coefficient of TiO2molecule in the physical adsorption stage in 600 ℃ in close to 100 times than that of 500 ℃. And the local charge distribution of ELF(0.750) of O-Ga1 in 500 ℃ is the most. The covalent bond of O-Ga1 bond is the strongest in 500 ℃. So the optimal temperature of TiO2adsorption on GaN (0001) is 500 ℃.
TiO2; GaN(0001);temperature; dynamics simulates;adsorption
1001-9731(2016)10-10128-06
国家自然科学基金资助项目(51172150); 四川省科技厅资助项目(2014JY0091)
2015-11-20
2016-03-09 通讯作者:梁晓琴,E-mail: lxqygr@163.com
梁晓琴 (1973-),女,四川南江人,教授,博士,主要从事材料理论计算研究。
O484; TB383
A
10.3969/j.issn.1001-9731.2016.10.023
