克-雅氏病5例临床分析
2016-11-12郎文娟孙元元冯加纯
郎文娟, 孙元元, 李 慧, 王 栋, 崔 俐, 冯加纯
克-雅氏病5例临床分析
郎文娟,孙元元,李慧,王栋,崔俐,冯加纯
目的提高对克-雅氏病(Creutzfeldt-Jakob disease,CJD)的认识及重视。方法回顾性分析5例散发性CJD的临床资料。结果本组亚急性起病2例,慢性起病3例。主要的临床症状和体征有进行性痴呆、精神行为异常、共济失调、肌阵挛、头晕、锥体外系征、锥体束征、言语笨拙、癫痫等。头部MRI弥散加权像3例表现为基底节区病变,4例表现为皮质异常信号。脑电图均有异常,2例存在背景脑电α节律解体,4例发作间期出现较多尖慢波,1例发作间期出现欠规则三相波。4例脑脊液14-3-3蛋白为阳性。结论CJD多为亚急性或慢性起病,病程进展迅速,以快速进行性痴呆为特征临床表现,头部MRI、脑电图、脑脊液14-3-3蛋白为主要辅助检查。
CJD;进行性痴呆;头部MRI;脑电图;14-3-3蛋白
克-雅氏病(Creutzfeldt-Jakob disease,CJD)又称为皮质-纹状体-脊髓变性,是由变异的朊蛋白引起的中枢神经系统致死性进展性疾病。其发病率低,病死率高,早期临床诊断困难,现总结我院近2年收治的5例CJD的临床特点,旨在提高临床医生对此病的认识及重视。
1 资料与方法
1.1研究对象选择我院2013年7月~2015年6月收治的5例散发性CJD患者,女性4例,男性1例,年龄56~71岁,平均62岁。5例患者中农民4例,干部1例,均无手术史,无CJD家族史。
1.2研究方法详细采集所有患者的病史并行体格检查,总结其临床表现(见表1),全部患者均行头部MRI、脑电图检查,血清、脑脊液送至中国疾病预防控制中心病毒病预防控制所-病毒病预防控制所朊病毒病室行14-3-3蛋白及基因检测,出院后进行随访。
2 结 果
2.1临床表现亚急性起病2例,慢性起病3例。首发症状头晕2例,记忆力减退2例,言语笨拙1例。主要的临床症状和体征:进行性痴呆5例,精神行为异常5例,共济失调3例,肌阵挛1例,头晕2例,锥体外系征4例,锥体束征1例,畏光1例,复视1例,言语笨拙2例,癫痫发作1例。
2.2血清与脑脊液检查5例患者均行血清基因及脑脊液14-3-3蛋白检测。4例脑脊液14-3-3蛋白检测阳性(western-blot 法),1例阴性;血液PRNP基因序列分析均为:(1)与标准序列对比出现序列无突变。(2)129 位氨基酸多态性为M/M 型。(3) 219 位氨基酸多态性为E/E 型。5例患者的脑脊液压力都正常(分别为100、145、100、120、180 mmH2O),常规生化中,1例白细胞稍高9×106/L(正常8×106/L),1例蛋白轻微增高0.49g/L(正常0.15~0.45 g/L),2例葡萄糖增高4.26、6.62 mmol/L(正常3.9~6.1 mmol/L),氯化物均正常。脑脊液梅毒螺旋体特异抗体、快速血浆反应素实验、结核抗体IgG、TORCH病毒抗体均为阴性。2例行血及脑脊液边缘性脑炎抗体检测,结果为阴性。边缘性脑炎抗体包括: 抗N-甲基-D-天冬氨酸受体(NMDA-R-Ab)、接触素相关蛋白2(CASPR2-Ab) 、α 氨基羟甲基恶唑丙酸受体1(AMPA1-R-Ab)、α-氨基羟甲基恶唑丙酸受体2(AMPA2-R-Ab)、γ-氨基丁酸B1 亚单位受体抗体(GABA2-R-Ab)、富亮氨酸胶质瘤失活1 自身抗体(LGI1-Ab)。血及脑脊液副肿瘤检测指标阴性。3 例患者行血清肿瘤标志物检测,1例神经元特异性烯醇化酶(NSE)增高26.81 ng/ml(正常<25.00 ng/ml)。血清肿瘤标志物包括:鳞状细胞癌抗原,癌胚抗原,糖类抗原15-3、72-4、19-9、125,甲胎蛋白,细胞角蛋白19 片段,神经元特异性烯醇化酶。
2.3辅助检查5例患者均行头部MRI及24h长程脑电图。头部MRI(见图1~图3):3例出现双侧尾状核、壳核异常信号,表现为T1W呈稍低或稍高信号,T2W、Flair呈稍高信号,DWI呈高信号;4例出现皮质异常信号(1例右侧颞顶枕叶、左侧顶枕叶,1例双侧额顶枕叶、右侧颞叶部分皮质,1例左侧额颞顶枕叶,1例双侧额顶枕叶、左侧颞叶),表现为T1W呈等或稍低信号,T2W、Flair呈等或高信号,DWI呈高信号。脑电图:2例背景脑电α节律解体,4例发作间期单侧或双侧颞区、额区枕区见较多尖慢波,1例发作间期右侧额区见欠规则三相波,部分呈PLED现象,1例发作间期左侧半球阵发性尖化的δ 波活动,呈周期性改变。1例监测到临床及脑电发作,4例全脑功能下降。
2.4临床诊断5例均诊断为很可能CJD。
2.5治疗及预后入院后给予改善循环、改善脑代谢、营养神经及营养支持治疗,病情进一步加重。随访:2例在1 m内死亡;1例健在至今7 m,较住院时未见明显变化;1例健在至今11 m,发病后10 m记忆力丧失;1例失访。

表1 临床资料
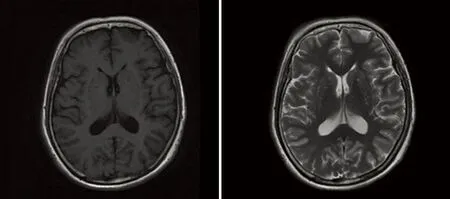
图1双侧额顶枕叶、左侧颞叶T1W呈稍低信号,T2W呈高信号

图2 双侧额顶枕叶、左侧颞叶Flair、DWI序列呈高信号
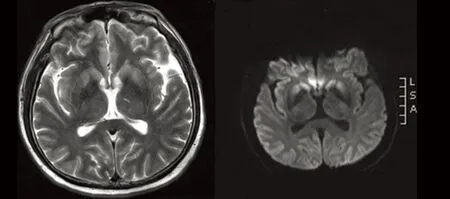
图3 双侧尾状核、壳核T2W、DWI序列呈高信号
3 讨 论
CJD是目前已明确的人类朊蛋白病中最常见的类型,是由Creutzfeldt和Jakob于1920年和1921年先后报道。本病好发于50~70岁人群,男女均可发病,受感染后的潜伏期为4~30 y[1]。根据不同病因,临床上将其分为:散发型、遗传型、医源型、新变异型4种类型。其中散发型CJD最常见,占85%,其病因不清;遗传型占5%~15%,是由于PrP基因突变所致;医源型是由于医院内行诊断或治疗时使用被污染的组织或器械所致;变异型与食用患牛海绵状脑病的肉类有关[2]。本文5例患者PRNP基因序列分析无突变,为散发型CJD。在2014 年中国克雅氏病监测网络病例特征分析中[3],310 份血液样本进行PRNP基因检测,其中297 例129 位多态性为M/M纯合子,占总病例数的95. 81%,294 例219 位多态性为E/E 纯合子,占总病例数的94. 84%,本文5例患者129 位为M/M 型,219 位为E/E 型,符合我国129 位、219 位等位基因分布特征。文献报道,129 位纯合子比杂合子更易患sCJD[4,5],219 位等位基因纯合子对CJD 可能是一种保护因素[6,7]。
CJD多为亚急性或慢性起病,病程呈快速进行性发展。主要临床表现为皮层功能损害、锥体外系、小脑功能障碍、脊髓前角损害和锥体束损害[1]。Rabinovici等[8]对114例散发克-雅氏病患者的首发症状和来自法国、英国、日本三个群体的此类患者的早期症状进行研究,其中认知障碍是最常见的首发症状,占40%,小脑症状、体质方面的症状、行为异常各占20%[8]。大约1/3的患者有早期体质方面的症状,有时被认为是该病的前驱症状,部分症状很难在解剖学上分类,包括疲乏、衰弱无力、头痛、心神不安、头晕、睡眠改变、饮食改变和不能解释的体重下降[8,9]。行为异常在早期症状中占20%~30%,在半数患者中贯穿疾病整个过程[9],皮质症状(如失语、失用、注意力降低、计算力缺失)在早期症状中占15%,也有一半患者整个疾病过程中都存在这一症状[8,9]。7%~17%的患者早期可出现视觉症状和动眼神经功能障碍,30%~40%发生在临床进程中[8,9]。约5%~10%的患者有首发和早期的感觉症状[8]。本文患者中,首发症状头晕2例,记忆力减退2例,言语笨拙1例。主要的临床症状和体征有进行性痴呆5例,精神行为异常5例,共济失调3例,肌阵挛1例,头晕2例,锥体外系征4例,锥体束征1例,畏光1例,复视1例,言语笨拙2例,癫痫发作1例。
CJD主要的辅助检查包括脑脊液检查、头部MRI、脑电图。脑电图作为无创性检查,患者易接受,在疾病中晚期出现的周期性同步放电(Periodic synchronous discharge,PSD)是诊断CJD的重要依据,且与肌阵挛有紧密联系,但对早期诊断不敏感。在CJD发病早期,EEG的基本节律解体,背景脑波不断慢化,之后,在慢波背景上出现周期性发作波,不久周期性发作波渐渐消失,慢波背景更加严重,这三个阶段存在的时间因病程的长短而异[10]。有研究报道[10],周期性发作波在病程的6~ 17 w(平均11. 8 w)出现,周期性发作波与检查的时间和频度有关。本文2例存在背景脑电α节律解体,4例发作间期出现较多尖慢波,1例发作间期出现欠规则三相波,部分呈PLED现象,5例患者不同程度上符合CJD脑电图特征,其中出现不规则PSD的患者出现了肌阵挛。5例中仅有1例出现三相波,考虑与其余4例检查时间过早或过晚有关,且均未复查脑电图。
目前普遍认为头部MRI是诊断CJD的重要影像学检查。其特异性和敏感性较CT高,患者早期头部CT检查无明显异常,头部MRI检查为早期诊断CJD提供了重要影像学依据。近几年研究表明,弥散加权像(diffusion-weighted imaging,DWI)对于CJD的早期诊断有很高敏感性(92.3%)和特异性(93.8%)[11]。早期CJD患者即可在DWI上出现皮质和(或)基底节区异常高信号;疾病晚期可消失。张家堂等[12]认为对于CJD患者,DWI 异常信号与临床表现及脑电图PSD有较高的一致性,并且DWI 异常信号较临床症状和体征以及PSD 表现更早、更敏感。曹笃等[13]研究与此观点相似。因此,对临床可疑的CJD 患者,DWI 检查完全可以作为早期、无创性、准确诊断CJD 的重要手段。Zerr等[14]提出头部MRI诊断依据:壳核和尾核或至少两个脑皮质(颞、顶、枕叶)在FLAIR或者DWI上呈现高信号。本文3例头部MRI均表现为基底节区病变,4例表现为皮质异常信号,出现“花边征”,5例患者DWI均出现异常信号。
脑脊液的一般检查除了少数人有轻微蛋白升高,可无明显异常。CSF14-3-3蛋白对诊断CJD有较大意义,但对CJD诊断的敏感性和特异性存在争议。以往多数研究认为其对CJD诊断的敏感性和特异性分别达93%~97%、84%~100%[15],有学者提出质疑,Geschwind等[16]研究认为敏感性只有53%。14-3-3蛋白阳性还可见于急性脑梗死、病毒性脑炎、CO中毒、副肿瘤综合征等疾病[17]。2012年美国神经病学会建议当强烈怀疑克雅病而诊断尚未明确时,应检测14-3-3蛋白[18]。14-3-3蛋白的检出与发病至检测时的病程有关,病程越短,检出率越高,,动态复查脑脊液14-3-3蛋白有助于CJD的诊断与鉴别[16]。此外,一些研究发现S100β、NSE、tau蛋白或者这些标志物的结合比14-3-3蛋白单独检测有更高的准确性[19,20]。2015年在对克雅病和非朊蛋白快速进展的痴呆患者的研究中发现tau蛋白比14-3-3蛋白或NSE诊断上更准确,但是DWI/ADC MRI比所有的生物标记物更加准确[19]。本文4例患者14-3-3蛋白为阳性,且介具有符合CJD的临床表现,因此对诊断此病有很大意义。
一个最新的检测方法,应用实时诱导转换(RT-QuIC)将脑脊液中朊蛋白放大成淀粉样纤维来检测,具有 77%~92%的敏感性,同时具有更高的特异性(99%~100%)[20]。当应用于取自被感染者的鼻粘膜时,这个检查在诊断上可能有更高的准确性[20],尽管这需要在大量非朊蛋白快速进展的痴呆患者中重复,但RT-QuIC是一个非常有希望的生前诊断方法。
CJD的诊断需临床表现与辅助检查相结合,参考2009年欧洲磁共振-克雅病联盟推荐标准[14]:(1)临床症状:进行性痴呆、视力障碍或小脑症状、锥体或锥体外系症状、无动缄默;(2)辅助检查:脑电图尖慢复合波(PSWC)、脑脊液14-3-3蛋白阳性(病程在2 y以内)、典型的头部MRI表现[壳核和尾核或至少两个脑皮质(颞、顶、枕叶)在FLAIR或DWI上呈现高信号]。符合(1)中2项和(2)中至少一项诊断很可能CJD,符合(1)中2项并且病程少于2 y者诊断可能CJD,若脑活检发现海绵状变和PrPSC者可确诊CJD。此5例患者均未做活检证实,根据诊断标准,5例均诊断为很可能CJD。
在临床工作中,遇到迅速进展的痴呆患者,应高度怀疑CJD,此病需与帕金森病、阿尔兹海默病、OPCA、遗传性进行性舞蹈病等相鉴别,尽快行腰穿、头部MRI弥散加权像、脑电图检查,必要时复查。目前本病尚无有效的治疗方法,正常细胞型朊蛋白转变为致病型朊蛋白是致病的关键环节,因此可作为治疗的主要靶点。目前的研究热点主要有药物疗法、免疫疗法、RNA干扰等。随着医学进步,相信在不久的将来,我们能找到治疗此病的有效方法。
[1]吴江. 神经病学. 第2版[M]. 北京:人民卫生出版社,2010. 213.
[2]林世和,张丽. Creutzfeldt-Jakob病不同类型的特点[J]. 中华神经科杂志,2012,45(2):76-77.
[3]肖康,周伟,张宝云,等. 2014年中国克雅氏病监测网络病例特征分析[J]. 疾病监测,2016,1:18-23.
[4]侯星生,高晨,张宝云 ,等. 中国不同民族人群中PrP蛋白基因第129位氨基酸多态性分析[J]. 中华实验和临床病毒学杂志,2002,2:5-8.
[5]Chen C,Wang JC,Shi Q,et al. Analyses of the survival time and the influencing factors of chinese patients with prion diseases based on the surveillance data from 2008-2011[J]. PLos One,2013,8(5):e62553.
[6]Shibuya S,Higuchi J,Shin RW,et al. Protective prion protein polymorphisms against sporadic Creutzfeldt-Jakob disease[J]. Lancet,1998,351(9100):419.
[7]Kobayashi A,Teruya K,Matsuura Y,et al. The influence of PRNP polymorphisms on human prion disease susceptibility: an update[J]. Acta Neuropathol,2015, 130(2):159-170.
[8]Rabinovici GD,Wang PN,Levin J,et al. First symptom in sporadic Creutzfeldt-Jakob disease[J]. Neurology,2006,66(2):286-287.
[9]Brown P,Cathala F,Castaigne P,et al. Creutzfeldt-Jakob disease:clinical analysis ofa consecutive series of 230 neuropathologically verified cases[J]. Ann Neurol,1986,20(5):597-602.
[10]逯恒东,陈芷若,李乐加. Creutzfeldt-Jakob病的脑电图改变[J]. 临床神经病学杂志,2001,6:359-360.
[11]Shiga Y,Miyazawa K,Sato S. Diffusion-weighted MRI abnormalitiesas an early diagnostic marker for Creutzfeldt-Jakob disease[J]. Neurology,2004,63(3):443-449.
[12]张家堂,蒲传强,贾渭泉,等. Creutzfeldt-Jakob病磁共振弥散加权像与临床表现及脑电图一致性的研究[J]. 中华神经医学杂志,2006,2:188-191.
[13]曹笃,魏有东,李琦,等. 磁共振弥散加权对克-雅氏病早期诊断的意义[J]. 中风与神经疾病杂志,2012,29(8):726-728.[14]Zerr I,Kallenberg K,Summers DM,et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease[J]. Brain,2009,132(10):2659-2668.
[15]于雪凡,林世和,赵珩. 脑脊液14-3-3蛋白对不同类型Creutzfeldt-Jakob病的诊断价值[J]. 临床神经病学杂志,2006,1:77-78.[16]Geschwind MD,Martindale J,Mifier D,et al. Challenging the clinicalutility of the 14-3-3 protein for the diagnosis of Sporadic Creutzfeldt-Jacob disease[J]. Arch Neurol,2003,60:813-816
[17]孙瑞红,林世和. 脑脊液中特殊蛋白质的检测与Creutzfeldt-Jakob病[J]. 临床神经病学杂志,2007,20:234.
[18]Muayqil T,Gronseth G,Camicioli R. Evidence-based guideline:diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease:report of the guideline development subcommittee ofthe American Academy of Neurology[J]. Neurology,2012,79(14):1499-1506.
[19]Forner SA,Takada LT,Bettcher BM,et al. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt-Jakobdisease[J]. Neurol Clin Pract,2015,5(2):116-125.
[20]Kim MO,Geschwind MD. Clinical update of Jakob-Creutzfeldt disease[J]. Curr Opin Neurol,2015,28(3):302-310.
5 case clinical analysis of sporadic Creutzfeldt-Jakob disease
LANGWenjuan,SUNYuanyuan,LIHui,etal.
(NeuroscienceCenter,DepartmentofNeurology,JilinUniversityFirstHospital,Changchun130021,China)
ObjectiveTo improve the awareness and attention of Creutzfeldt-Jakob Disease(CJD). MethodsThe clinical data of 5 patients with sCJD were analyzed retrospectively. Results2 cases were subacute onset,3 cases were chronic onset. The major clinical symptoms and physical signs of CJD were progressive dementia,mental behavior,ataxia and myoclonus,dizziness,extrapyramidal signs,pyramidal signs,speech clumsy,epilepsy,etc. Among 5 case MRI diffusion-weighing pictures,3 cases showed abnormal signal in basal part,4 cases showed abnormal signal in cortices. The EEG results were all abnormal,2 cases showed the disintegration of the α rhythm on background of EEG,4 cases showed much sharp slow wave complex in interictal,1cases showed atypical triphasic wave in interictal. 4 cases CSF 14-3-3 protein tests showed positive. ConclusionAlmost all of sCJDs are subacute or chronic onset. It progress rapidly and is characterized by rapid progressive dementia. MRI、EEG and CSF 14-3-3 protein test are the major auxiliary examination for CJD.
CJD;Progressive dementia;MRI;EEG;14-3-3 protein
1003-2754(2016)05-0429-04
2016-01-03;
2016-03-25
(吉林大学白求恩第一医院神经内科,神经科学中心,吉林 长春 130021)
冯加纯,E-mail:fengjcfrank@qq.com
R51
A
