Hutchinson-Gilford早老综合征
2016-11-06张韡苏忠兰吴侃宋昊温斯健杨莹刘白林志淼孙建方
张韡 苏忠兰 吴侃 宋昊 温斯健 杨莹 刘白 林志淼 孙建方
·研究报道·
Hutchinson-Gilford早老综合征
张韡 苏忠兰 吴侃 宋昊 温斯健 杨莹 刘白 林志淼 孙建方
报告1例Hutchinson-Gilford早老综合征(HGPS)。对1例患儿及其父母外周血LMNA基因11号外显子和侧翼序列进行测序。患者男,5岁,全身皮肤呈硬皮病样改变,生长迟滞,特殊面容,毛发稀少。髋、膝关节均不能完全伸直,呈“骑马样站姿”。患儿LMNA基因1l号外显子c.1824C>T杂合点突变,父母均未检测到该位点突变。文中还通过回顾性分析,探讨中国人群中通过基因学诊断的18例病例的疾病特点。我国基因学诊断的18例HGPS中,9例经典型HGPS均为散发病例,基因表型均上出现c.1824C>T杂合突变。患儿均在1岁以内发病,出生时基本未表现出“异常”。患儿男女性别比例为2∶1,以男孩受累明显;非经典型患儿基本在家族内发病,男女受累情况类似,3个家庭中均发现c.1579C>T纯合突变。
早衰;DNA突变分析;分子诊断技术;遗传现象;基因,LMNA
Hutchinson-Gilford早老综合征(Hutchinson-Gilford progeria syndrome,HGPS;OMIM176670) 是一种累及皮肤、脂肪、肌肉、骨骼、血管及心脏多器官系统,具有特殊面容及临床表现的少见遗传病[1]。现报道我们诊治的1例HGPS。
1 临床资料
1.1 病历资料:患儿男,5岁。出生后6周因双下肢硬肿2周就诊。病程中无发热,精神好,大小便无异常。初发时未予重视,其后下肢硬肿范围逐渐扩大,渐及躯干部而就诊。体检:心肺腹未见异常。腹壁、背部及双下肢皮肤肿胀变硬,静脉显露。以硬肿症收入当地儿童医院治疗,给予营养支持,改善循环等治疗无效而出院。其后反复于当地多家医院诊治,检查血尿粪常规及血生化无异常,细胞免疫及体液免疫水平无明显异常,甲状腺功能正常,组织病理检查(图1):表皮轻度增厚,皮突稍下延,基底层色素增加,真皮及皮下脂肪层广泛胶原纤维及成纤维细胞增生,部分胶原纤维呈嗜酸性均质化改变,经治疗无效而到我院就诊。患儿系足月顺产,父母非近亲结婚,否认家族遗传病史。出生时无异常,母乳喂养。1岁8个月时身体发育情况评价显示:身高79.4 cm,体重8.35 kg,身体发育水平低下。智力发育正常。
体检:患儿身材短小,面容特殊:头大面小、双眼及前额突出,鼻大致正常,声音尖细。头皮毛细血管扩张,呈网状分布,周围皮肤硬化,毛发稀少,前囟未闭。全身皮肤萎缩变硬,呈暗红色,表面高低不平可触及硬块。两臀部相连,阴囊明显肿胀变硬。双下肢对称,但髋、膝关节均不能完全伸直,呈“骑马样站姿”(图 2)。
作者单位:210042 南京,中国医学科学院 北京协和医学院 皮肤病研究所病理科(张韡、吴侃、宋昊、温斯健、杨莹、刘白、孙建方);江苏省人民医院皮肤科(苏忠兰);北京大学第一医院皮肤科(林志淼)
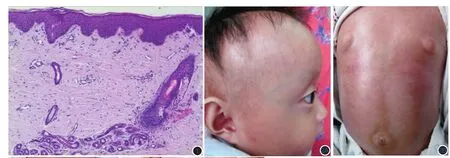

图1 皮损组织病理:表皮轻度增厚,皮突稍下延,基底层色素增加,真皮及皮下脂肪层广泛胶原纤维及成纤维细胞增生,部分胶原纤维呈嗜酸性均质化改变(HE×100) 图2 患儿的临床表现2A:头大面小,前额突出,毛发稀少 2B:腹部皮肤萎缩变硬,呈暗红色,表面高低不平可触及硬块;2C:两侧臀部相连;2D:阴囊明显肿胀变硬,髋、膝关节均不能完全伸直
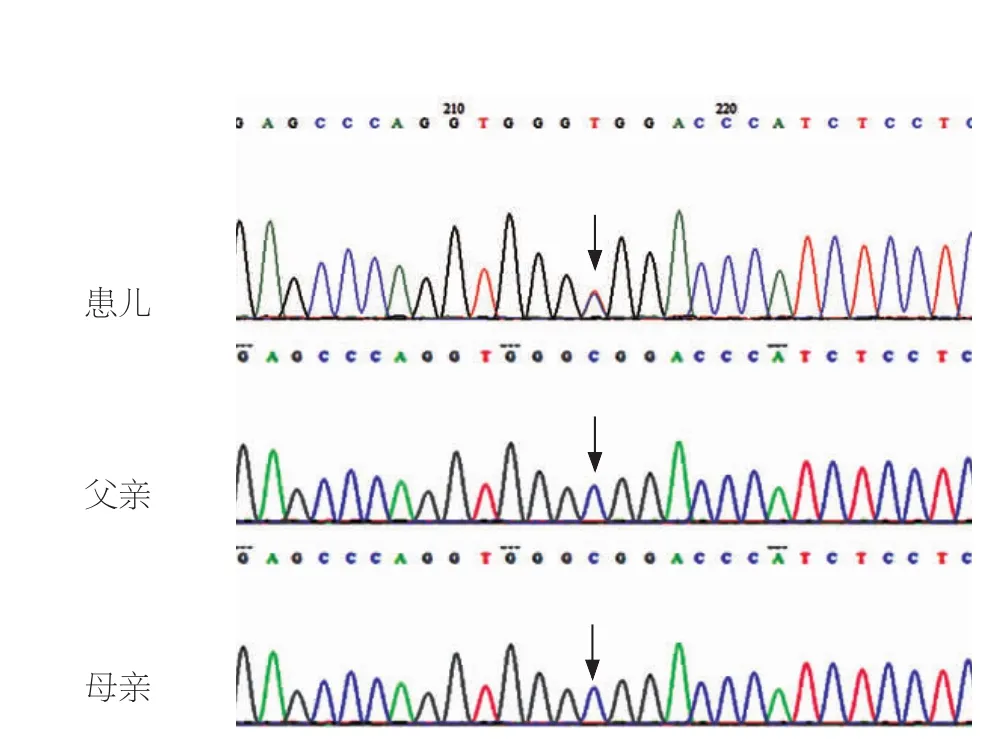
图3 患儿LMNA基因的c.1824C>T杂合突变测序图及患儿父母的LMNA基因测序图 患儿LMNA致病基因发生c.1824C>T杂合突变
1.2 基因学检测:提取患儿及其父母外周血DNA,采用PCR及Sanger测序检测患者LMNA致病基因外显子及侧翼序列,发现LMNA致病基因发生c.1824C>T杂合突变,导致其编码蛋白出现剪切位点改变,其父母及200例健康对照的外周血DNA检测未发现该突变位点(图3)。所有结果经过双向测序验证。
1.3 临床诊断:HGPS。患儿随访4年,随访期间每年行心血管B超未见异常,智力发育良好,四肢肿胀情况略有好转。目前暂无特效治疗,继续随访。
2 讨论
HGPS是Hutchinson最早于1886年报道,其发病率为1∶4 000 000~1∶8 000 000,患者常死于心肌梗死或脑卒中等心脑血管疾病,平均寿命为13岁[2]。该病罕见,迄今中国人群中通过基因学仅确诊18例[3-9]。9例经典型HGPS均为散发病例,基因表型上出现c.1824C>T杂合突变。患儿发病均在1岁以内,且出生时通常未表现出“异常”。患儿男女性别比例为2∶1,以男孩受累明显;非经典型患儿基本在家族内发病,男女受累情况类似,3个家庭中均发现c.1579C>T纯合突变,另外,还有散发1例患儿出现IVS6+1G>T基因突变,1例患儿出现c.1318G>A纯合突变。由LMNA基因及其编码蛋白Lamin A/C异常引起的一组人类遗传病被称为核纤层蛋白病。核纤层蛋白病的种类多,临床表型相似,包括:经典型HGPS、非经典型HGPS、下颌骨-肢端发育不良A型、限制性皮肤病、Werner综合征及脂肪营养不良、胰岛素抵抗型糖尿病、弥漫性白黑皮病样丘疹、肝脂肪变性和心肌病综合征等10余种疾病。一个基因,多种疾病[10]。
根据HGPS的临床表型及基因改变,HGPS可分为经典型和非经典型[1-2]。经典型HGPS遗传方式为散发、常染色体显性遗传,且90%与LMNA基因杂合突变c.1824C>T相关。经典型HGPS表现为出生时正常,1岁内逐渐出现多系统的临床状:典型面部特征,包括:头大面小,双眼突出,鼻梁窄鼻尖高突,声音尖细。头皮血管扩张,毛发稀少;全身皮肤萎缩变硬,表面高低不平可触及硬块;髋、膝、踝等大关节均不能完全伸直,呈“骑马样站姿”;生长、发育迟滞等多系统生长障碍。随年龄增长,早老样外貌逐渐加重,面部特征将更加典型。本病发病初期在临床上呈现硬皮病样表现,病理上亦可看到真皮及皮下脂肪层广泛胶原纤维及成纤维细胞增生,但患儿往往有多系统的临床症状,且面部特征及站姿具有特征性,可以鉴别;另外,此病在组织病理上与纤维瘤病相类似,但根据患儿临床表现亦可鉴别。
[1]Korf B.Hutchinson-Gilford progeria syndrome,aging,and the nuclear lamina[J].N Engl J Med,2008,358(6):552-555.DOI:10.1056/NEJMp0800071.
[2]Pollex RL,Hegele RA.Hutchinson-Gilford progeria syndrome[J].Clin Genet,2004,66 (5):375-381.DOI:10.1111/j.1399-0004.2004.00315.x.
[3]Liang L,Zhang H,Gu X.Homozygous LMNA mutation R527C in atypical Hutchinson-Gilford progeria syndrome:evidence for autosomal recessive inheritance[J].Acta Paediatr,2009,98(8):1365-1368.DOI:10.1111/j.1651-2227.2009.01324.x.
[4]Zhang H,Chen X,Guo Y,et al.Hutchinson-Gilford progeria syndrome:report of 2 cases and a novel LMNA mutation of HGPS in China [J].J Am Acad Dermatol,2013,69 (4):175-176.DOI:10.1016/j.jaad.2011.07.002.
[5]Xiong Z,Lu Y,Xue J,et al.Hutchinson-Gilford progeria syndrome accompanied by severe skeletal abnormalities in two Chinese siblings:two case reports[J].J Med Case Rep,2013,7:63.DOI:10.1186/1752-1947-7-63.
[6]沈鉴东,尹飞,梁德生,等.罕见Hutchinson-Gilford早老综合征基因诊断一例 [J].中华医学杂志,2009,89(42):3022-3023.DOI:10.3760/cma.j.issn.0376-2491.2009.42.021.
[7]阳芳,李乾,郑利雄,等.早老综合征LMNA基因突变研究[J].中华皮肤科杂志,2014,47(7):465-468.DOI:10.3760/cma.j.issn.0412-4030.2014.07.004.
[8]Chu Y,Xu ZG,Xu Z,et al.Hutchinson-Gilford progeria syndrome caused by an LMNA mutation:a case report [J].Pediatr Dermatol,2015,32 (2):271-275.DOI:10.1111/pde.12406.
[9]覃霞,罗彦彦,袁广之,等.一个儿童早老症家系临床特征分析和致病基因研究[J].中华皮肤科杂志,2015,48(03):184-186.DOI:10.3760/cma.j.issn.0412-4030.2015.03.011.
[10]宋书娟,章远志,Zhong N.核纤层蛋白病——一个基因,多种疾病[J].北京大学学报(医学版),2005,37(1):96-99.
Hutchinson-Gilford progeria syndrome
Zhang Wei*,Su Zhonglan,Wu Kan,Song Hao,Wen Sijian,Yang Ying,Liu Bai,Lin Zhimiao,Sun Jianfang.*Department of Pathology,Institute of Dermatology,Chinese Academy of Medical Sciences and Peking Union Medical College,Nanjing 210042,China
To report a case of Hutchinson-Gilford progeria syndrome (HGPS).Peripheral blood samples were collected from a 5-year-old boy with HGPS and his parents.DNA was extracted from these samples,and PCR was performed to amplify exon 11 of the LMNA gene and its flanking sequences followed by DNA sequencing.The patient presented with scleroderma-like skin changes all over the body,growth retardation,distinctive facial features and hypotrichosis.His hip and knee joints could not be straightened completely,giving a horse-riding stance.A heterozygous mutation C.1824C > T was identified in exon 11 of the LMNA gene in the patient but not in either of his parents.A retrospective analysis was carried out on 18 Chinese patients with genetically diagnosed HPGS.Of them,9 classical cases were all sporadic with a heterozygous mutation of C.1824C>T.None of the patients with classical HPGS showed abnormality at birth,but all of them developed symptoms within 1 year after birth.Boys were more frequent to be affected by classical HPGS than girls,with the male/female ratio being 2:1.There was a familial tendency for the occurrence of atypical HPGS,and boys and girls appeared to be affected by HPGS at a similar probability.Three families with atypical HPGS all showed a homozygous LMNA mutation c.1579C>T.
Progeria;DNA mutational analysis;Molecular diagnostic techniques;Genetic phenomena;Genes,LMNA
s:Sun Jianfang,Email:fangmin5758@aliyun.com;Lin Zhimiao,Email:zhimiaolin@bjmu.edu.cn
2015-06-18)
10.3760/cma.j.issn.1673-4173.2016.02.001
孙建方,Email:fangmin5758@aliyun.com;林志淼,Email:zhimiaolin@bjmu.edu.cn
