离子液体/季铵盐辅助氯过氧化物酶促氧化合成聚酚
2016-11-03王圣洁刘丽霞蒋育澄胡满成李淑妮翟全国
王圣洁, 刘丽霞, 蒋育澄,2, 胡满成,2, 李淑妮,2, 翟全国,2
(1. 陕西师范大学化学化工学院, 2. 大分子科学陕西省重点实验室, 西安 710119)
离子液体/季铵盐辅助氯过氧化物酶促氧化合成聚酚
王圣洁1, 刘丽霞1, 蒋育澄1,2, 胡满成1,2, 李淑妮1,2, 翟全国1,2
(1. 陕西师范大学化学化工学院, 2. 大分子科学陕西省重点实验室, 西安 710119)
基于氯过氧化物酶(CPO)催化氧化苯酚衍生物单体, 建立了一个聚酚的绿色合成体系. 以对苯基苯酚、 对甲基苯酚、 4-乙基苯酚、 对羟基肉桂酸、 对异丙基苯酚和邻甲基苯酚等6种底物为考察对象, 以聚合物的产率、 聚合度及热稳定性为评价指标, 研究了体系中引入离子液体(ILs)或季铵盐(QAS)以及底物结构和反应微环境等对聚合反应和聚合物性质的影响. 结果表明, 引入少量咪唑类ILs或QAS可有效提高产物收率, 其中ILs/QAS的阳离子基团越大和疏水链越短, 越有利于酶催化聚合反应的进行; 而ILs/QAS添加量的影响则呈现“钟罩”型规律. 同时, 苯酚对位取代远比邻位取代有利于聚合反应进行; 而对位取代基中烷基类给电子基团比芳香基取代更有优势, 所得聚合物的聚合度和热稳定性相对增大, 但随着取代基团的增大, 其空间位阻不利于聚合物产率的提高; 反应体系的pH应控制在弱酸性至近中性, 以避免竞争性的副反应的发生; 而氧化剂H2O2则需要采用间歇式加入以抑制瞬时过浓导致CPO活性中心卟啉环的氧化损伤. 基于CPO的活性中心结构分析了聚合机理.
氯过氧化物酶; 聚苯酚; 离子液体/季铵盐; 底物结构; 反应微环境
酚类聚合物中以酚醛树脂的应用最为广泛, 可用于制造各种塑料、 涂料、 胶粘剂及合成纤维等[1~5]. 这类材料由苯酚与甲醛聚合而成, 但由于甲醛的毒性使其生产和使用都受到限制[1], 所以人们一直在寻求尽量少用或者避免使用甲醛制备酚醛树脂的合成方法或者可以替代酚醛树脂的聚酚产品, 以解决传统工业生产中的污染问题.
近年来, 人们发现一些过氧化物酶能够催化H2O2氧化苯酚或使其衍生物单体聚合, 所获得的聚酚热稳定性较好, 有替代酚醛树脂的潜力. 由于酶促反应条件温和, 环境友好, 过程易控, 选择性好等优势引起了人们的极大兴趣, 其中研究最多的是将辣根过氧化物酶[6~8]和漆酶[9,10]用于聚酚合成. 但这些酶促聚合反应都是在水溶液中进行, 受有机底物水溶性的制约, 使聚合物的产率较低, 通常需要加入有机溶剂作为共溶剂. 然而大多数酶是亲水性的, 有机溶剂会使酶分子表面的水化层被破环而导致活性受损, 甚至完全失活, 同时有机溶剂的引入也不利于反应的绿色化.
本文采用氯过氧化物酶(CPO)催化H2O2氧化5种酚类衍生物单体聚合, 制备了相应的聚酚, 通过引入少量咪唑类离子液体或季铵盐可有效提高单体的聚合效率并提高聚合物的热稳定性. 所选用的CPO兼具过氧化物酶、 过氧化氢酶和细胞色素P-450的催化功能, 目前被认为是血红素过氧化物酶家族中催化活性最广泛的酶[11~18]. 反应过程一步完成, 绿色环保; 所得聚合物纯度较高, 反应中所需的酶用量极少.
1 实验部分
1.1试剂与仪器
各种酚衍生物包括对苯基苯酚、 对甲基苯酚、 4-乙基苯酚、 对羟基肉桂酸、 对异丙基苯酚和邻甲基苯酚; 咪唑类离子液体:溴化1-乙基-3-甲基咪唑([EMIM][Br])、 溴化1-丙基-3-甲基咪唑([PMIM]·[Br])、 溴化1-丁基-3-甲基咪唑([BMIM][Br]); 季铵盐包括四甲基溴化铵(TMABr)、 四乙基溴化铵(TEABr)、 四丙基溴化铵(TPABr)、 四丁基溴化铵(TBABr). 上述试剂均购自国药集团化学试剂有限公司. 十二烷基苯磺酸钠(SDBS)、 30%(质量分数)过氧化氢及其它化学试剂均购自西安化学试剂厂. 以上试剂均为分析纯, 所有溶液均以超纯水配置(R>18.25 MΩ·cm).
真菌Caldariomycesfumago的培养及CPO的提纯参照Hager等[19]的方法, 但在酶的提纯阶段用丙酮代替甲醇进行沉淀. 将所提取的酶溶液浓缩至7.4 mg/mL, 置于4 ℃冷柜保存. Monochlorodimedon(MCD)氯化反应分析表明CPO氯化活性为5700 U/mL; 2,2′-联氯-二(3-乙基-苯并噻唑-6-磺酸)(ABTS)氧化反应分析表明CPO氧化活性为3400 U/mL; CPO纯度达到Rz=1.09(Rz=A398/A280=1.44, 视为纯酶).
EQUINX55型傅里叶变换红外光谱仪(德国Brucher公司); UV-1700型紫外-可见分光光度计(岛津国际贸易上海有限公司); Q50热分析系统(美国TA公司); Agilent1100-Esquire6000型液相色谱-质谱联用仪(LC-MS, 德国布鲁克公司); Q600 SDT热重差热联用分析仪, 美国TA公司; Q1000 DSC型热重分析仪.
1.2实验过程
向100 mL样品管中依次加入50 mL 0.1 mol/L磷酸缓冲液(pH= 5.5)和酚衍生物底物(终浓度达到0.1 mol/L), 用恒温磁力搅拌器在25 ℃及固定转速下持续搅拌. 然后加入CPO(终浓度达到1.5×10-7mol/L), 聚合反应用H2O2启动, 采用间歇式加入H2O2, 每10 min滴加一次5%的双氧水, 每次滴加8 μL, 共滴加500 μL, 12 h后终止反应. 将产物离心20 min后真空抽滤, 用50%(体积分数)的乙醇洗涤以除去过量的底物、 表面活性剂以及少量的CPO和H2O2, 于70 ℃下烘干得到粉末状产物. 聚合物的产率根据下式计算:
引入ILs或QAS时步骤同上, 其中ILs(25 ℃时液态存在)的添加量为5%(体积分数), QAS(25 ℃时固态存在)的添加量达到终浓度0.08 mol/L.
红外光谱表征采用KBr压片. 热分析温度范围:-180~725 ℃; 灵敏度:0.2 μW; 加热速率:0.01~200 ℃/min; 温度范围:室温~1500 ℃; 灵敏度:0.1 μg. 产物用N,N-二甲基甲酰胺(DMF)溶解后测定紫外-可见光谱, 扫描范围:200~900 nm. 样品以甲醇溶解后以Agilent 1100-Esquire 6000 LC-MS分析检测其聚合度.
2 结果与讨论
2.1产物表征
5种苯酚衍生物的结构如图1所示.
Fig.1 Phenols compounds used in the polymerization catalyzed by CPO(A) p-Methyl phenol; (B) p-ethyl phenol; (C) p-propyl phenol; (D) p-phenyl phenol; (E) p-hydrox-cinnamic acid.
2.1.1聚合物FTIR分析以对苯基苯酚及对甲基苯酚的反应产物为例, 其CPO催化聚合反应产物的红外光谱如图2所示. 其中3450~3490 cm-1的特征吸收峰归属为酚羟基的伸缩振动, 3100 cm-1的特征吸收峰归属为饱和烷烃的碳氢伸缩振动, 1600~1400 cm-1的特征吸收峰归属为苯环骨架的伸缩振动, 1100 cm-1的特征吸收峰归属为碳氧键的伸缩振动. 4-乙基苯酚、 对羟基肉桂酸和对异丙基苯酚的反应产物的红外图谱也同样表明:CPO催化H2O2氧化苯酚衍生物的产物为聚酚, 即这5种酚类衍生物单体都发生了聚合.
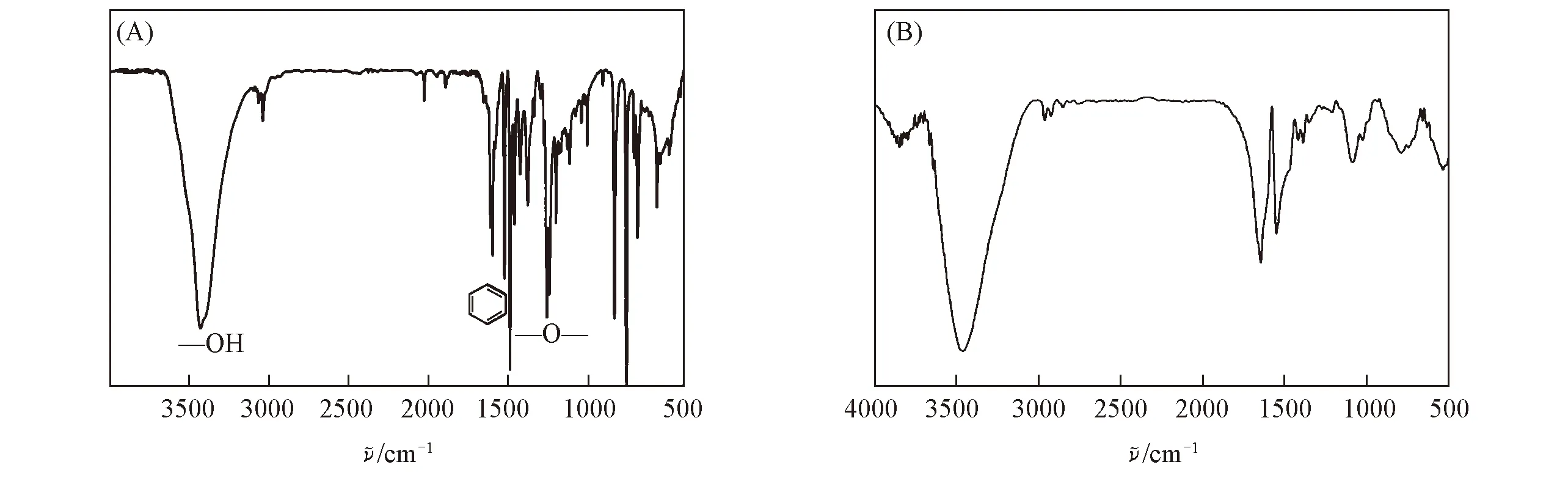
Fig.2 FTIR spectra of phenyl polymers(A) Polymer of p-phenyl phenol; (B) polymer of p-methyl phenol.
通过对比实验表明, 如果反应体系中仅加入氧化剂H2O2, 无CPO存在, 则没有聚合物出现, 这说明酚衍生物聚合归因于CPO的催化.
2.1.2液相色谱-质谱联用采用液相色谱-质谱联用分析检测聚合物的聚合度, 根据质谱图中的分子离子峰的质荷比(m/z值)来测定单体的聚合数.
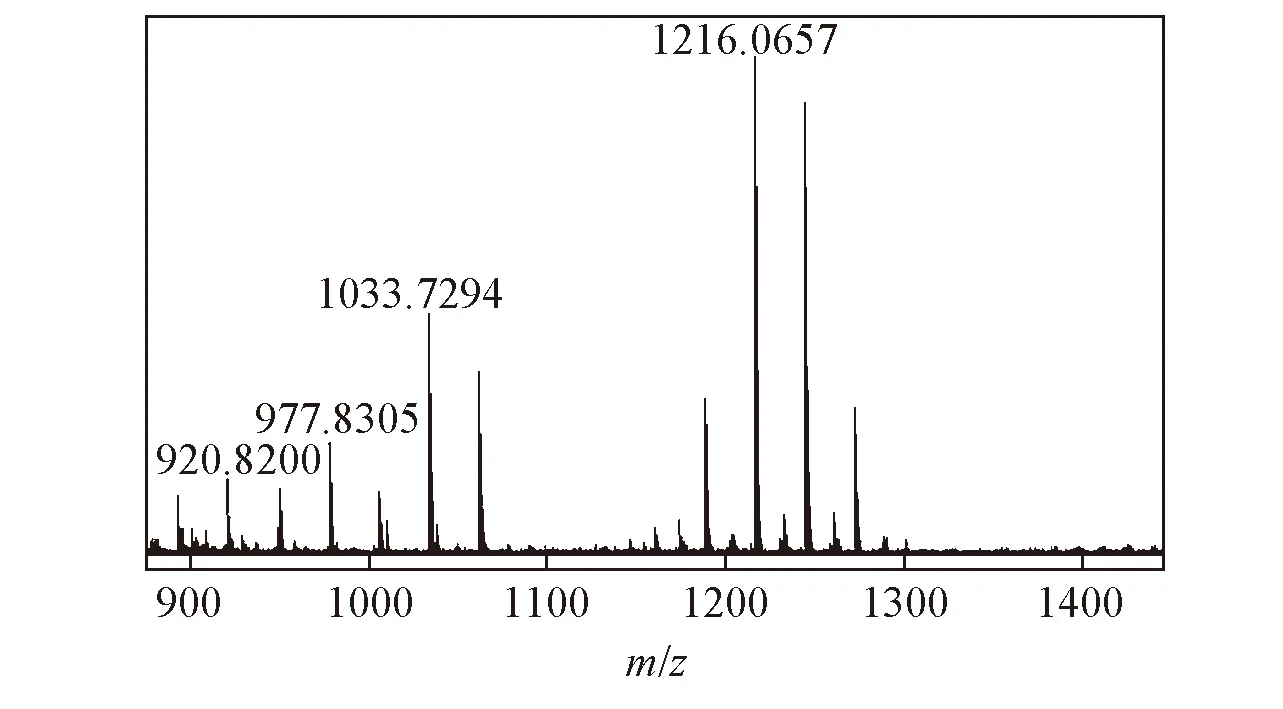
Fig.3 Mass spectrum of the product of CPO-catalyzed oxidation of p-phenyl phenol
图3为对苯基苯酚的聚合产物的质谱图. 根据质谱检测的结果, 对苯基苯酚、 对甲基苯酚、 4-乙基苯酚、 对异丙基苯酚、 对羟基肉桂酸等5种酚衍生物的单体聚合数分别可以达到4~7, 7~8, 10~13, 13~14以及3~4.
2.1.3热分析差示扫描量热分析及热重分析表明所获得的聚酚都表现出较好的热稳定性, 分解温度都在150 ℃以上. 以不同底物为单体制备的聚酚的热分解温度分别为对苯基苯酚:170 ℃; 对甲基苯醛:160 ℃; 4-乙基苯酚:155 ℃; 对羟基肉桂酸 120 ℃. 同时空气气氛中对甲基苯酚和对羟基肉桂酸的聚合物分别在575 ℃和600 ℃完全分解, 而在氮气气氛中超过1000 ℃时仍分别有30%和17%的聚合物存在.
2.2离子液体/季铵盐对酚衍生物聚合反应的影响
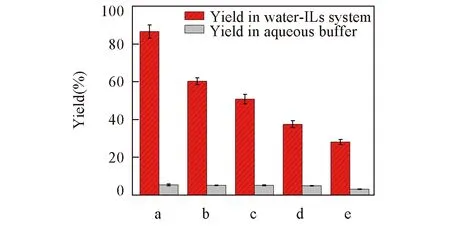
Fig.4 Comparison of different phenols polymerization catalyzed by CPO in aqueous buffer in the presence of 0.08×10-3 mol/L TMABr to that in pure buffer aqueousa. p-Phenyl phenol; b. p-hydroxy-cinnamic acid; c. p-methyl phenol; d. p-ethyl phenol; e. p-propyl phenol.
由于受底物水溶性的制约, CPO催化H2O2氧化酚衍生物单体在水溶液中聚合的产率很低. 本文引入少量离子液体或季铵盐可大大提高目标产物的产率. 对苯基苯酚、 对羟基肉桂酸、 对甲基苯酚、 4-乙基苯酚、 对异丙基苯酚等5种酚衍生物聚合物的产率(图4)可分别提高81%(5.3%→86.5%), 55%(5.1%→60.22%), 45%(5%→50.67%), 32%(4.8%→37.4%)和25%(3%→28%).
离子液体与季铵盐都具有“两亲”性结构, 即同时拥有亲水性的“头基”和疏水性的“尾”链(烃基), 因此在离子液体/季铵盐的“介导”下, 亲水性的CPO分子与疏水性的底物之间能够有效接触, 这是聚合物产率提高的原因之一; 此外, 反应体系中有离子液体/季铵盐存在时, 聚合物能够更好地成链并生长; 同时, 与有机溶剂相比, 两亲性的离子液体/季铵盐对酶蛋白分子表面水化层的破环导致的酶活性损失也小得多, 因而酶分子对离子液体/季铵盐的耐受性更强, 使其与有机溶剂相比较在反应体系中可以添加至较高的比例[20,21].
本文所选用的咪唑类离子液体包括溴化1-乙基-3-甲基咪唑([EMIM][Br])、 溴化1-丙基-3-甲基咪唑([PMIM][Br])、 溴化1-丁基-3-甲基咪唑([BMIM][Br]). 季铵盐包括四甲基溴化铵(TMABr)、 四乙基溴化铵(TEABr)、 四丙基溴化铵(TPABr)和四丁基溴化铵(TBABr)等. 阴离子皆为Br-而不是Cl-, 这是为了避免Cl-在CPO催化的氧化反应中与酚衍生物底物形成竞争.
聚酚的产率也受ILs/QAS添加量的影响. 本文在2%~20%的离子液体和0.02~0.2 mol/L季铵盐的范围内考察了添加剂含量的影响, 结果如图5所示(以对苯基苯酚为例). 总体来看, QAS的效果要优于ILs, 但两者都呈现“钟罩”型的变化趋势. 反应初期, 随着添加剂含量的增加, 聚合物产率迅速增加, 到达一个极值后开始降低, 表明此时CPO的活性开始受损. 引入0.08 mol/L TMABr的反应体系中聚酚的产率可达到最大值86.5%. 在没有酶存在的ILs/QAS体系中对苯基苯酚未发生聚合.
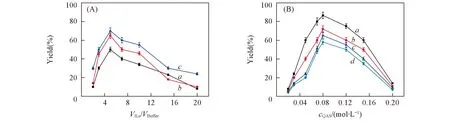
Fig.5 Effect of ILs/QAS content on the p-phenylphenol polymerization (A) Imidazolium ILs. a. [EMIM][Br], b. [PMIM][Br], c. [BMIM][Br]; (B) QAS. a. TMABr, b. TEABr, c. TPABr, d. TBABr.
同时ILs/QAS的结构也会影响底物的聚合产率. 离子液体中阳离子烃基链的长短影响着酶蛋白分子的稳定性, 较短的烃基链更有利于蛋白的二级结构的稳定. 本文聚合物的产率随着ILs/QAS阳离子烃基链的增长而下降. 其中烃基链最短的季铵盐四甲基溴化铵(TMABr)的效果最好, 反应体系中引入TMABr时可获得最大的聚合物产率(86.5%).
但ILS或QAS加入前后, 聚合物的聚合度基本上没有发生变化.
2.3反应微环境的影响
对反应体系的pH在2.0~6.5范围内进行了考察. 因为CPO在弱酸性的环境中比较稳定, pH>6.0时酶基本失活; 同时pH<2.5时CPO活性中心的酸碱催化组分Glu183处于质子化的状态, 会增加反应体系中阴离子的竞争反应从而影响目标产物的生成. 由图6可以看出, 聚合反应适合的pH范围为5.5~6.0.

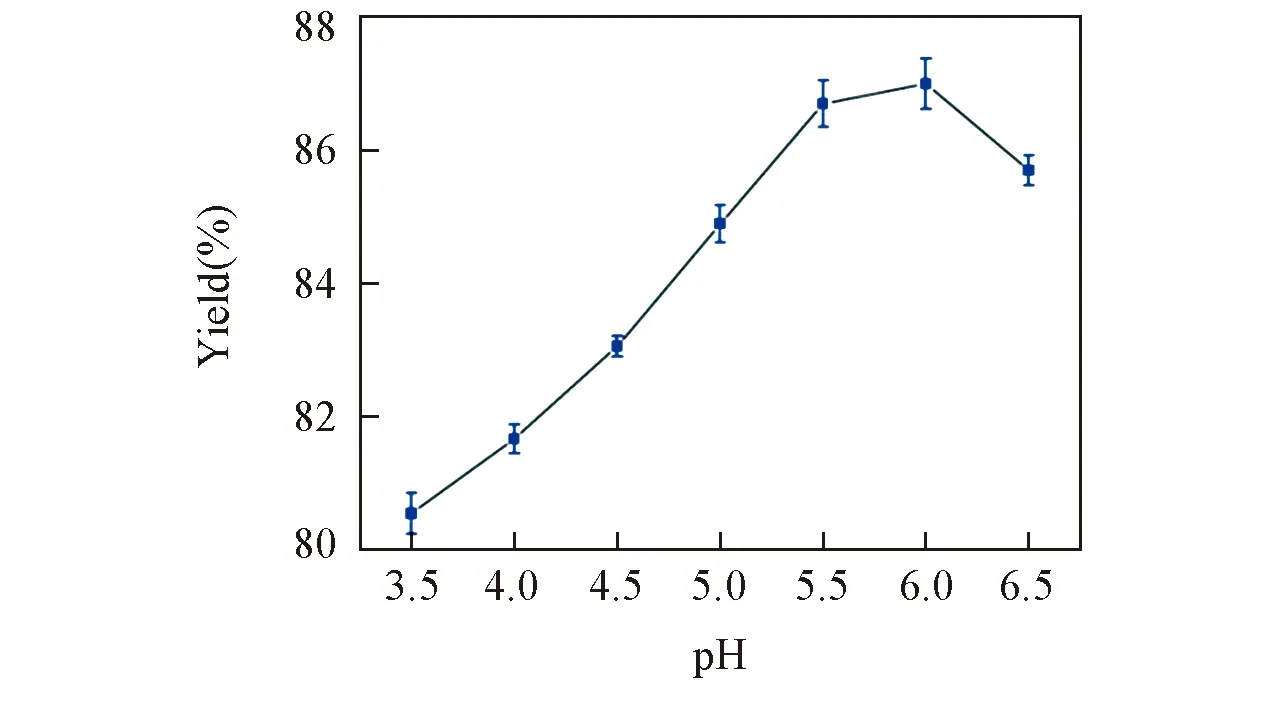
Fig.6 Influence of pH on p-phenyl phenol polymerization in aqueous buffer in the presence of 0.08×10-3 mol/L TMABr
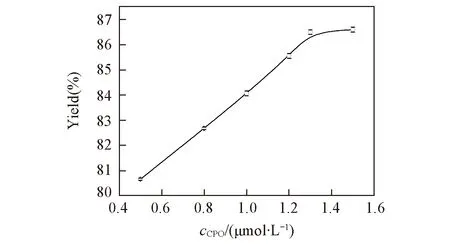
Fig.7 Effect of amount of enzyme on poly phenyl phenol
聚合物的产率随CPO的用量的增大迅速提高, 但最终趋于平稳(图7, 以对苯基苯酚聚合为例). 若考虑最大收率的, 同时保证CPO的用量最少, 则所需的酶用量在(1.34~1.61)×10-7mol/L的范围内, 表明聚合反应的酶用量极少, 极具应用潜力.
2.4底物结构对聚合度的影响
底物结构与聚合度关系密切. 根据液质分析的结果, 对苯基苯酚、 对甲基苯酚、 4-乙基苯酚、 对异丙基苯酚、 对羟基肉桂酸等5种酚衍生物的单体聚合数分别达到4~7, 7~8, 10~13, 13~14以及3~4, 说明羟基对位的给电子基团比吸电子基团更有利于底物聚合. 同时, 给电子基团的烃基链越长, 聚合度也越高, 而吸电子基团的情况则正好相反, 随着吸电子基团烃基链的增长聚合度下降. 本文还考察和比较了邻位取代的甲基苯酚与对位取代的甲基苯酚的聚合情况, 发现邻甲基苯酚几乎不发生聚合.
虽然酚衍生物的酶催化聚合反应也可在有机溶剂-缓冲溶液中进行, 如二氧六环、 丙酮以及DMF等, 但有机溶剂的加入对酶活性有所抑制, 所需要的酶用量较大; 同时有机溶剂的大量使用也不符合绿色化学的要求[24,25]; 在缓冲溶液中加入水溶性的模板剂, 如聚乙二醇(PEG), 可以得到聚合度较高的酚聚合产物, 但将模板剂与聚合产物分离很困难, 而且, 使用模板剂后酶用量并没有明显下降[26]. 而本文中采用ILS/QAS-缓冲溶液体系酶促合成聚酚不仅提供了一条绿色合成路线, 而且CPO酶用量极少.
2.5CPO催化的苯酚衍生物聚合机理
Scheme 1是以对乙基苯酚的聚合为例阐明底物的自由基聚合机理.
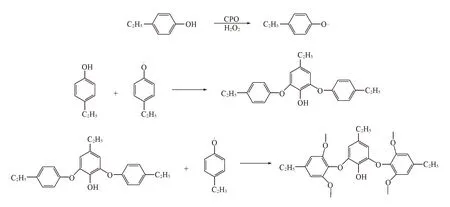
Scheme 1 Polymerization of phenol through oxidation catalyzed by CPO
CPO活性中心结构非常独特, 其血红素的“近端”与细胞色素P-450相似, 是以半胱氨酸(Cys29)的巯基硫原子作为铁的第五轴向配体, 而在其它的过氧化物酶中占据这一位置的是组氨酸的咪唑氮原子; 但与P-450不同的是CPO分子中血红素的“远端”是亲水性环境; 同时CPO的血红素活性中心并没有暴露在酶蛋白分子表面, 底物不易直接接近CPO的血红素边缘, 需要先通过一个疏水性通道才能到达活性中心, 但这个通道对底物的大小有限制, 即碳原子数超过9的直链烃无法通过底物通道[27]. 这一结构特点提示聚合反应不可能在CPO活性中心完成(否则聚合产物无法穿过底物通道进入溶液), 因此推测聚合过程是一个自由基反应, 首先CPO催化H2O2氧化酚衍生物单体产生自由基, 生成的自由基经过底物通道从活性中心进入溶液主体, 并完成聚合过程.
3 结 论
采用CPO催化H2O2氧化苯酚衍生物单体制备聚酚, 通过引入少量的咪唑类离子液体或季铵盐可有效提高聚合物的产率并改善聚合物的热稳定性, 其中以反应体系引入0.08 mol/L的季铵盐四甲基溴化铵(TMABr)的效果最好, 产率可提高80%以上(产率由不加季铵盐的5%提高到86.5%), 且产物的热稳定性优于纯水相合成的聚酚; 对位取代苯酚远比邻位取代有利于聚合反应, 且对位取代基中烷基类给电子基团相比芳香基取代更有优势, 但取代基的空间位阻不利于聚合反应. 聚合反应属于自由基机理, 聚合过程是在溶液主体中而并非在CPO活性中心完成. 酶促制备过程一步完成, 产物提纯简便, 反应温和高效, 环境友好, 且酶用量极少.
[1]Guo L. Y., Zhang B., Bai S. Y., Ma X. Y., Wang Z. M.,E-Polymers, 2015, 15, 195—201
[2]Raj M. M., Raj L. M., Shah T. B., Patel P. M.,J.Therm.Anal.Calorim., 2010, 101, 1003—1009
[3]Shafizadeh J. E., Guionnet S., Tillman M. S., Seferis J. C.,J.Appl.Polym.Sci., 1999, 73, 505—514
[4]Wang W. B., Zhao Z. Y., Gao Z. H., Guo M. G.,BioResources, 2012, 7, 1972—1983
[5]Greenblatt M.,Lab.Med., 1988, 19(7), 425—428
[6]Eker B., Zagorevski D., Zhu G. Y., Linhardt R. J., Dordick J. S.,J.Mol.Catal.B:Enzym., 2009, 59, 177—184
[7]Shogren R. L., Willett J. L., Biswas A.,Carbohydr.Polym., 2009, 75, 189—191
[8]Reihmann M. H., Ritter H.,Macromol.Biosci., 2001, 1, 85—90
[9]Shumakovich G., Otrokhov G., Vasil′eva I., Pankratov D., Morozova O., Yaropolov A.,J.Mol.Catal.B:Enzym., 2012, 81, 66—68
[10]Tan Y., Deng W. F., Li Y. Y., Huang Z., Meng Y., Xie Q. J., Ma M., Yao S. Z.,J.Phy.Chem.B, 2010, 114(15), 5016—5024
[11]Zhang R., He Q., Chatfield D., Wang X.,Biochem., 2013, 52(21), 3688—3701
[12]Wang Y. L., Nie Y. Y., Jiang Y. C., Hu M. C., Li S. N., Zhai Q. G.,Chem.J.ChineseUniversities, 2012, 33(6), 1344—1349(王亚丽, 聂艳艳, 蒋育澄, 胡满成, 李淑妮, 翟全国. 高等学校化学学报, 2012, 33(6), 1344—1349)
[13]Nie Y. Y., Jiang Y. C., Hu M. C., Li S. N., Zhai Q. G.,ActaChimicaSinica, 2010, 68(10), 982—988(聂艳艳, 蒋育澄, 胡满成, 李淑妮, 翟全国. 化学学报, 2010, 68(10), 982—988)
[14]Manoj K. M.,BBA-ProteinsProteom, 2006, 1764, 1325—1339
[15]Sundaramoorthy M., Terner J., Poulos T. L.,Structure(London), 1995, 3, 1367—1377
[16]Sundaramoorthy M., Terner J., Poulos T. L. S.,Chem.Biol., 1998, 5, 461—473
[17]Longoria A. M., Hu H. L., Vazquez-Duhalt R.,Appl.Biochem.Biotechnol., 2010, 162, 927—934
[18]Roman P., Cruz-Silva R., Vazquez-Duhalt R.,SyntheticMet., 2012, 162, 794—799
[19]Morris D. R., Hager L. P.,J.Biol.Chem., 1966, 241(8), 1763—1768
[20]Jan R., Rather G. M., Bhat M. A.,J.Solution.Chem., 2014, 43, 685—695
[21]Gorke J., Srienc F., Kazlauskas R.,Biotechnol.Bioproc.E., 2010, 15, 40—53
[22]Jin R. X., Li C. N., Zhi L. F., Jiang Y. C., Hu. M. C., Li. S. N., Zhai Q. G.,Carbohydr.Res., 2013, 370, 72—75
[23]Sheng X., Horner J. H., Newcomb M.,J.Am.Chem.Soc., 2008, 130, 13310—13320
[24]Dordick J. S., Marletta M. A., Klibanov A. M.,Biotechnol.Bioeng., 1987, 30, 31—36
[25]Akkara J. A., Senecal K. J., Kaplan D. L.,J.Polym.Sci.:PartA,Polym.Chem., 1991, 29, 1561—1547
[26]Kim Y. J., Uyama H., Kobayashi S.,Macromolecules, 2003, 36, 5058—5060
[27]Hager L. P.,J.Biol.Chem., 2010, 285, 14852—14860
(Ed.:D, Z)
† Supported by the National Natural Science Foundation of China(No.21176150) and the Fundamental Research Funds for the Central Universities of China(No.GK201505007).
Enzymatic Polymerization of Phenols Catalyzed by Chloroperoxidase in the Presence of Ionic Liquids/Quaternary Ammonium Salts†
WANG Shengjie1, LIU Lixia1, JIANG Yucheng1,2*, HU Mancheng1,2,LI Shuni1,2, ZHAI Quanguo1,2
(1. School of Chemistry & Chemical Engineering,2.KeyLaboratoryofMacromolecularScienceofShaanxiProvince,ShaanxiNormalUniversity,Xi’an710119,China)
A green enzymatic approach for the synthesis of phenol polymersfrom substituted phenols monomer by chloroperoxidase(CPO)-catalyzed H2O2-oxidation was proposed in this paper. The yield and thermal stabi-lity of polymers ofp-methyl phenol,p-ethyl phenol,p-propyl phenol,p-phenyl phenol andp-hydroxy-cinnamic acid were studied based on the presence of imidazolium-based ionic liquids(ILs) or quaternary ammonium salts(QAS), the effect of structure of the substrates and the reaction microenvironment. The results showed that the introduction of little amount of ILs/QAS can improve the production of polymerization of phenols efficiently, in which ILs/QAS with bigger cation group and shorter hydrophobic chain was much more effective, while the influence of ILs/QAS amount on polymerization of phenols showed a “ball type” pattern. Moreover, it was found thatp-substituted phenol and electron-donating group were more beneficial to the increase of yield and thermal stability of phenol polymers compared too-substituted phenol and electron-withdrawing group. However, the steric hindrance was increased with the increasing size of substituent group, which was not beneficial to the polymerization of phenols. The pH value should be controlled as weak acid or even near-neutral to avoid competitive side reaction, while the adding of H2O2should be in a batch type to suppress the oxidative damage of heme caused by the instantaneous concentrated H2O2. The mechanism of polymerization was also analyzed and proposed based on the characteristics of structure of CPO active site.
Chloroperoxidase; Polyphenol; Ionic liquid/Quaternary ammonium salt; Structure of substrate; Reaction microenvironment
10.7503/cjcu20160236
2016-04-13. 网络出版日期:2016-08-23.
国家自然科学基金(批准号:21176150)和中央高校专项基金(批准号:GK201505007)资助.
O632.7
A
联系人简介:蒋育澄, 女, 博士, 教授, 主要从事酶工程研究. E-mail:jyc@snnu.edu.cn
