肺结核治疗的新方向——抗结核药物的肺部递送
2016-10-12谌茜
谌茜
摘 要 本文结合肺部的解剖学和生理学特征以及肺结核的病理学特征重点阐释了抗结核药物肺部递送的优势和应关注的问题。通过近几年若干肺部递送抗结核药物的研究实例说明了肺结核治疗的这一新途径的可行性。
关键词 肺结核 肺部递送 巨噬细胞靶向 空气动力学直径
中图分类号:R943; R521 文献标识码:A 文章编号:1006-1533(2016)17-0071-07
A new direction for tuberculosis treatment— the pulmonary delivery of anti-tuberculosis drugs
CHEN Xi*(Sichuan Nursing Vocational College, Chengdu 610100, China)
ABSTRACT This review highlights the advantages of the pulmonary delivery of anti-TB agents and the associated concerns based on the understanding of the characteristics of both the anatomy and physiology of the lung and the TB pathology. The feasibility of this new drug delivery approach has been recently proven by some case-studies on the pulmonary delivery of anti-TB agents.
KEY WORDS tuberculosis; pulmonary delivery; macrophage targeting; aerodynamic diameter
肺结核是一种由空气中结核分枝杆菌引起的细胞内感染性疾病,是一种慢性传染性疾病。目前肺结核的治疗仍呈现巨大的挑战,一方面是由于过长的治疗周期,另一方面是复杂的多种药物疗法带来的不可预知的不良反应。将抗结核药物通过感染途径直接递送至呼吸系统似乎是一种较为合理的手段。它可避免药物的首过效应,降低药物导致的全身不良反应,也为提高药物对病灶的靶向作用创造了条件。
1 呼吸系统
1.1 呼吸系统的解剖学及生理学基础
呼吸系统可以被分为两个功能区域,即呼吸道和肺部。呼吸道由鼻腔,咽,喉,气管,支气管和各级支气管分支组成,是气体进出肺部的通道。这个部分类似于一棵树的树干和树枝。肺部则包含呼吸性细支气管,肺泡管,肺泡囊以及肺泡。呼吸道最远端的终端细支气管分支为呼吸性细支气管,其进一步细分为肺泡管,肺泡囊和肺泡。气体交换就发生在肺泡部分。与呼吸道相比,肺部更像是一棵树的叶子。
人体呼吸道的表面积约为2~3 m2,而肺泡的表面积则高达100 m2左右[1]。除了表面积上巨大差异外,两个主要的功能区域的上皮组织结构也存在着显著的差异。呼吸道包含自上而下由逐渐变细的上皮组织,人体支气管(human bronchi)上皮细胞直径约为3~5 mm,而终端细支气管(terminal bronchioles)的上皮细胞仅有0.5~1 mm[1],其上均覆盖着用于保护的黏液层(图1)。与此相比,肺泡(alveoli)则是由直径更小的单层上皮细胞组成。除了上皮组织直径上的明显差异外,由支气管至肺泡上的上皮组织厚度也从约70 μm骤降至0.1~0.2 μm,这同时也就表明由肺入血的生物学屏障在逐渐变薄[3]。
在肺泡中,单层的上皮组织主要由肺泡1型细胞组成,其约覆盖肺泡表面积的96%。除此之外,分布于肺泡囊中心的主要是肺泡1型母细胞及少量能产生肺部表面活性剂的肺泡2型细胞[4]。肺部表面活性剂构成了覆盖于肺泡上皮组织的内衬层。在气体交换的表面则由肺部巨噬细胞“巡逻”(每5亿个肺泡有12~14个巨噬细胞)[5],它们主要负责吞噬和消化异源颗粒、过多的表面活性剂、病毒和细菌[6]。
1.2 适于肺部递送颗粒的基础要求
吸入的颗粒在呼吸系统中沉降的位置主要基于颗粒的空气动力学直径,这一参数主要由颗粒的几何学直径、密度及形状决定[7]。图2直观地阐释了颗粒的空气动力学直径对其在呼吸道中沉降位置的影响。根据图中的曲线可以看出,通过制备不同空气动力学直径的颗粒可以有效地控制它们在呼吸道的沉降位置,从而达到理想的治疗效果。例如,如果药物颗粒需要被递送的肺泡区域(alveolar region),1~5 μm,特别是3 μm的颗粒是较为理想的。另一方面,如果药物的靶点在呼吸道(airways)中,空气动力学直径为5~10 μm的颗粒最为理想。
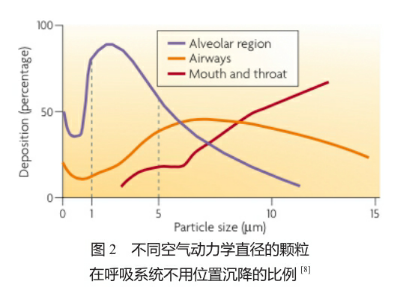
在设计吸入药物时,除了颗粒的沉降位置,颗粒的清除机制也应充分考虑。沉降在呼吸系统中的颗粒被清除的机制主要取决于其几何学直径、沉降位置和在呼吸系统液体中的溶解度[4]。无论是吸收还是非吸收的清除方式,不溶的吸入颗粒首先接触的是呼吸道中的黏液层或是肺泡中的内衬液层。尽管巨噬细胞也少量分布于呼吸道中[6,9],但在此区域中的不溶颗粒的主要清除方式是黏液纤毛清除[10]。这种清除方式可以保证大多数不溶颗粒在24~48 h内被清除[11-12]。然而一些纳米颗粒在沉降到呼吸道远端后,可能会通过那里较薄的黏液层从而避免被黏膜纤毛清除[9]。沉降到肺泡区域的不溶颗粒,其主要的清除方式是巨噬细胞的吞噬[13]。而巨噬细胞的摄取取决于颗粒大小。一般来说,1~3 μm的颗粒是最易被巨噬细胞识别和吞噬的[14-15]。那些更小的纳米颗粒可能主要与非吞噬的上皮细胞相作用[16],或穿过较薄的上皮细胞进入血液。之前有研究表明几何学直径为10~20 μm的多孔颗粒可以沉降在深肺区域并能有效地避免巨噬细胞的吞噬[17-18]。对于那些在一个轴线上大于20 μm的颗粒,如纳米纤维,太长而不能被巨噬细胞(细胞直径大多为15~20 μm)摄取。上述的这些不能被清除的颗粒可能会在肺中滞留数月或数年,甚至会引起一些非特异性的肺部炎症反应[19-20]。总的来看,基于不用的治疗目的,在设计吸入药物时均应考虑颗粒的沉降位置和清除方式。
2 肺结核
2.1 肺结核的流行病学
肺结核是由结核分枝杆菌引起的一种慢性传染性细菌疾病。根据联合国卫生组织在2013发布的全球结核病报告,2012年新增结核病人约860万,其中约130万死亡病例[21]。近些年,人类免疫缺陷病毒(human immunodeficiency virus, HIV)的传播促使了结核病的发展。2012年130万结核病死亡病例中就有30万是由艾滋病引发的[21]。除此之外,多重耐药菌株(multi-drug resistant strain, MDR strain),即至少对利福平和异烟肼两种一线抗结核药物耐药的菌株[22],和最近出现的广泛耐药菌株(extensively drug resistant strain, XDR strain), 即在多重耐药菌株的基础上对奎诺酮类抗生素和至少一种可注射的二线抗结核病药物耐药的新型菌株[23],使得肺结核的治疗变得更为复杂和困难。
2.2 肺结核的发病机制
结核分枝杆菌能够感染人体的很多器官,包括骨,脑膜,泌尿系统及皮肤。但据估计超过80%的结核分枝杆菌的感染发生在肺部[24],而且肺部的顶端是易感染部位。因此,肺结核的发病机制将在下面的内容中被重点讨论。
结核分枝杆菌在肺中的生命周期如图3所示。细菌的感染始于带有细菌的并且具有合适的空气动力学直径的液滴在肺部的沉降[26]。据估计最小的感染数量仅为1个结核分枝杆菌[25]。入侵的细菌进入肺泡部位后将会被肺泡巨噬细胞识别和吞噬。除了巨噬细胞,在感染的初始阶段体内专职的抗原呈递细胞(antigen presenting cells, APCs),如树突状细胞,也会与侵入的结核分枝杆菌相作用[27]。由于结核分枝杆菌的特殊性,被巨噬细胞吞噬的细菌不仅能有效地避免被溶酶体分解而且可以在巨噬细胞内复制新个体[28-29]。被感染的巨噬细胞会向上皮组织扩散,从而引起来自血管内单核细胞数量的增加。这些单核细胞与被感染的巨噬细胞一起形成了早期的感染灶(granuloma),这也是肺结核最明显的病理学特质。尽管对这一结构的认识尚不够明确,但有理由相信这是一种不同的免疫细胞(特别是T细胞和巨噬细胞)围绕被感染的巨噬细胞的特殊结构[30]。细菌在巨噬细胞内快速复制之后,感染灶将会被新形成的纤维结构包裹。与此同时,通过感染灶的血管数量骤减,使得结构内出现低氧状态从而抑制细菌的复制。随着空穴在感染灶内的增加,灶内开始逐渐塌陷坏死,结核分枝杆菌重新被释放到呼吸体统中,从而开始新的周期。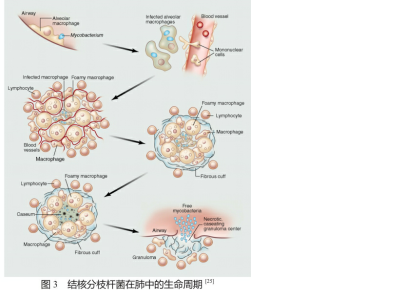
2.3 肺结核的现行治疗方式及限制
肺结核的治疗目标包括防止复发的治愈、传染源的控制及耐药性的预防[23,31]。为实现这些目标,长时间地服用多种抗结核药物时必须的。口服抗生素是治疗和控制肺结核的标准方式。目前一线抗结核病药物包括利福平(rifampicin, RIF)、异烟肼(isoniazid, INH)、吡嗪酰胺(pyrazinamide, PZA)和乙胺丁醇(ethambutol, EMB)。世界卫生组织(WHO)推荐的结核病治疗方式要求多重药物疗法[32],包括①在初始期的两个月每日服用RIF,INH,PZA和EMB;②在之后的四个月每日或每周三次服用RIF和INH;③对于多重耐药肺结核的治疗,WHO建议长达20个月服用多种一线及二线抗结核病药物[22]。如此长的治疗周期和复杂的疗法将会带来不可预知的不良反应及不理想的病人顺应性[33]。因此,对现行肺结核疗法的改进应聚焦到如何缩短治疗周期和降低药物不良反应上。
3 肺部递送药物在肺结核治疗中的优势
与肺部递送抗结核药物相比,口服需要经历胃肠道的降解和首过效应的代谢。尽管在口服和注射抗结核药物后,可达到理想的血药浓度,但这并不表示可将足够的药物量递送至病灶部位。为了到达作用靶点,抗结核药物必须从血管中转移至感染灶。在新生感染灶中血管量丰富,尚可为药物的递送提供充足的机会。但发展到后期的感染灶中血管量骤减,药物必须通过血液进入感染灶,之后扩散至空穴中,再穿过富含脂质的结核分枝杆菌细胞膜,最终与其作用靶点相结合(图4)。在后期的感染灶中存在着大量的停止复制的细菌,而匮乏的血流量却不能将足够的药物量递送至此。因此,选用合适的肺部药物递送体统不仅能在减少全身药物量的前提下实现较高的局部药物浓度[35-36],而且可以靶向作用于那些被细菌感染的巨噬细胞。
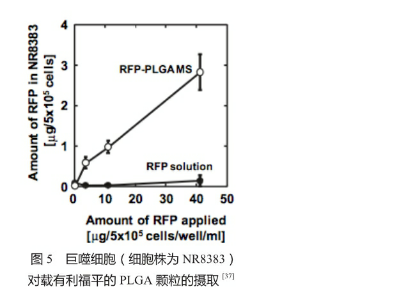
首先,靶向作用于巨噬细胞的前提是将载有药物的颗粒递送至感染部位(肺泡区域)。根据以前的结论,载有药物的颗粒应具有1~5 μm(最优为3 μm)的空气动力学直径才能保证可有效沉降于肺泡区域。其次,当颗粒沉降至肺泡区域之后,它们应较易被细菌感染的巨噬细胞识别并吞噬。几何学直径1~3 μm的颗粒较易被肺泡内的巨噬细胞吞噬。图5阐释了载有利福平的聚乳糖-羟基乙酸共聚物〔Poly(lactic-co-glycolic) acid, PLGA〕颗粒(平均直径为2 μm)被巨噬细胞吞噬的效率。数据显示,在相同的药物浓度下,利用颗粒包裹的药物递送系统相比于单纯的药物溶液对巨噬细胞具有更高的递送效率。有趣的是,有研究表明被感染的巨噬细胞吞噬颗粒的能力较未感染的巨噬细胞高[38],这为靶向递送抗结核药物提供了更大的可能。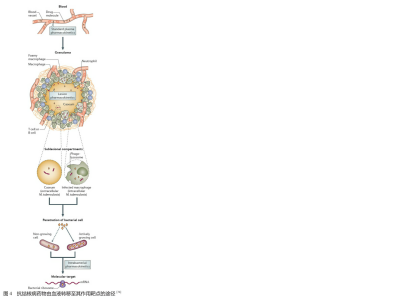
根据以上两点,为了实现肺部递送并靶向作用于巨噬细胞,载有药物的最适宜颗粒应该具有1~5 μm的空气动力学直径和1~3 μm的几何学直径。如果使用的辅料密度低于水的密度(1 g/cm3),如一些脂质材料,忽略其形状等因素的考虑,则颗粒的空气动力学直径可能会略小于其几何学直径,这与我们想要的结果是不一致的。在此种情况下应考虑一些特殊的颗粒制备技术,如喷雾干燥和冷冻干燥技术。另外,由于颗粒沉降后将会直接与水环境相接触,为确保药物能被巨噬细胞细胞吞噬,水不溶性材料可能更为合适。当然,除了颗粒大小和材质还有很多方面会影响巨噬细胞的选择性吞噬,文献在这方面做了很好的总结[39]。
4 用于治疗肺结核的吸入药物颗粒
近年来,通过肺部递送的方式来治疗肺结核引起了广泛的关注,特别是以干粉吸入剂的形式进行递送。用于肺部递送的粉末颗粒应具有良好的粉末学性质(如良好的粉末流动性),合适的空气动力学直径,并且能被肺部巨噬细胞选择性摄取。表1中列出了近4年来一些通过肺部递送方式来治疗肺结核的文献信息。
5 展望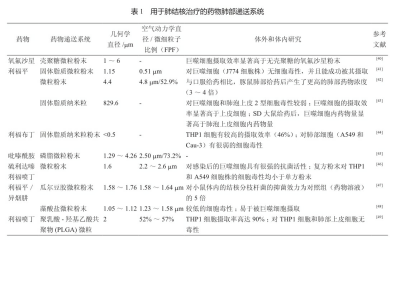
基于对呼吸系统生理学和肺结核病理学的认识,肺部递送抗结核药物为肺结核的治疗提供了新的途径。就制剂方面而言,为实现安全高效的治疗目的,选择合适的递送系统显得尤为重要。近几年来,对于递送颗粒的制备及辅料的选择,学术界已经进行了深入地研究,但剂型的临床前研究仍显不足。在未来的研究中,基于肺部递送抗结核药物的可行性、制剂技术及临床前研究的工作应给予更多的关注。
参考文献
[1] Patton JS. Mechanisms of macromolecule absorption by the lungs[J]. Adv Drug Deliver Rev, 1996, 19(1): 3-36.
[2] Patton JS, Byron PR. Inhaling medicines: delivering drugs to the body through the lungs[J]. Nat Rev Drug Discov, 2007, 6(1): 67-74.
[3] Courrier HM, Butz N, Vandamme TF. Pulmonary drug delivery systems: recent developments and prospects[J]. Crit Rev Ther Drug Carrier Syst, 2002, 19(4-5): 425-498.
[4] Weber S, Zimmer A, Pardeike J. Solid lipid nanoparticles(SLN) and nanostructured lipid carriers (NLC) for pulmonary application: a review of the state of the art[J]. Eur J Pharm Biopharm, 2014, 86(1): 7-22.
[5] Stone KC, Mercer RR, Gehr P, et al. Allometric relationships of cell numbers ?and size in the mammalian lung[J]. Am J Resp Cell Mol, 1992, 6(2): 235-243.
[6] Bur M, Henning A, Hein S, et al. Inhalative nanomedicineopportunities and ?challenges[J]. Inhal Toxicol, 2009, 21(S1): 137-143.
[7] Telko MJ, Hickey AJ. Dry powder inhaler formulation[J]. Resp Care, 2005, 50(9): 1209-1227.
[8] Byron PR. Prediction of drug residence times in regions of the human respiratory tract following aerosol inhalation[J]. J Pharma Sci-US, 1986, 75(5): 433-438.
[9] M?ller W, H?u?inger K, Winkler-Heil R, et al. Mucociliary and long-term particle clearance in the airways of healthy nonsmoker subjects[J]. J Appl Physiol, 2004, 97(6): 2200-2206.
[10] Lippmann M, Yeates DB, Albert RE. Deposition, retention, and clearance of inhaled particles[J]. Br J Ind Med, 1980, 37(4): 337-362.
[11] Newman SP, Agnew JE, Pavia D, et al. Inhaled aerosols: lung deposition and clinical applications[J]. Clin Phys Physiol Meas, 1982, 3(1): 1-20.
[12] Gonda I. Aerosols for delivery of therapeutic and diagnostic agents to the ?respiratory tract[J]. Crit Rev Ther Drug Carrier Syst, 1989, 6(4): 273-313.
[13] Sibille Y, Reynolds HY. Macrophages and polymorphonuclear neutrophils in lung defense and injury[J]. Am Rev Respir Dis, 1990, 141(2): 471-501.
[14] Chono S, Tanino T, Seki T, et al. Influence of particle size on drug delivery to rat alveolar macrophages following pulmonary administration of ciprofloxacin incorporated into liposomes[J]. J Drug Target, 2006, 14(8): 557-566.
[15] Lawlor C, OSullivan MP, Sivadas N, et al. The application of high-content analysis in the study of targeted particulate delivery systems for intracellular drug delivery to alveolar macrophages[J]. Mol Pharm, 2011, 8(4): 1100-1112.
[16] Yang W, Peters JI, Williams III RO. Inhaled nanoparticles—a current review[J]. Int J Pharm, 2008, 356(1): 239-247.
[17] Edwards DA, Ben-Jebria A, Langer R. Recent advances in pulmonary drug delivery using large, porous inhaled particles[J]. J Appl Physiol, 1998, 85(2): 379-385.
[18] Edwards DA, Dunbar C. Bioengineering of therapeutic aerosols[J]. Annu Rev Biomed Eng, 2002, 4(1): 93-107.
[19] Borm PJA, Kreyling W. Toxicological hazards of inhaled nanoparticles—potential implications for drug delivery[J]. J Nanosci Nanotechnol, 2004, 4(5): 521-531.
[20] Hoet PHM, Brüske-Hohlfeld I, Salata OV. Nanoparticles–known and unknown health risks[J/OL]. J Nanobiotechnol, 2004, 2-12. DOI: 10.1186/1477-3155-2-12.
[21] World Health Organization. Global tuberculosis report 2013[EB/OL]. [2016-07-08]. http://apps.who.int/iris/bitstre am/10665/91355/1/9789241564656_eng.pdf. ?
[22] Falzon D, Jaramillo E, Schünemann HJ, et al. WHO guidelines for the programmatic management of drug-resistant tuberculosis: 2011 update[J]. Eur J Hum Genet, 2011, 38(3): 516-528.
[23] Dube D, Agrawal GP, Vyas SP. Tuberculosis: from molecular pathogenesis to effective drug carrier design[J]. Drug Discov Today, 2012, 17(13-14): 760-773.
[24] Pandey R, Khuller GK. Antitubercular inhaled therapy: opportunities, progress and challenges[J]. J Antimicro Chemoth, 2005, 55(4): 430-435.
[25] Russell DG, Barry CE 3rd, Flynn JL. Tuberculosis: what we dont know can, and does, hurt us[J]. Science, 2010, 328(5980): 852-856.
[26] Kaufmann SHE. How can immunology contribute to the control of tuberculosis?[J]. Nat Rev Immunol, 2001, 1(1): 20-30.
[27] Wolf AJ, Linas B, Trevejo-Nu?ez GJ, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo[J]. J Immunol, 2007, 179(4): 2509-2519.
[28] Armstrong JA, Hart PDA. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes[J]. J Exp Med, 1971, 134(3): 713-740.
[29] Sinai AP, Joiner KA. Safe haven: the cell biology of nonfusogenic pathogen vacuoles[J]. Annu Rev Microbiol, 1997, 51(1): 415-462.
[30] Gordon S, Keshav S, Stein M. BCG-induced granuloma formation in murine tissues[J]. Immunobiology, 1994, 191(4): 369-377.
[31] Muttil P, Wang C, Hickey AJ. Inhaled drug delivery for tuberculosis therapy[J]. Pharm Res, 2009, 26(11): 2401-2416.
[32] World Health Organization. Fixed-dose combination tablets for the treatment of tuberculosis[EB/OL]. [2016-07-28] http:// apps.who.int/iris/bitstream/10665/65981/1/WHO_CDS_ CPC_TB_99.267.pdf.
[33] Prabakaran D, Singh P, Jaganathan KS, et al. Osmotically regulated asymmetric capsular systems for simultaneous sustained delivery of anti-tubercular drugs[J]. J Controlled Release, 2004, 95(2): 239-248.
[34] Dartois V. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells[J]. Nat Rev Microbiol, 2014, 12(3): 159-167.
[35] Zhou H, Zhang Y, Biggs DL, et al. Microparticle-based lung delivery of INH decreases INH metabolism and targets alveolar macrophages[J]. J Controlled Release, 2005, 107(2): 288-299.
[36] Du Toit LC, Pillay V, Danckwerts MP. Tuberculosis chemotherapy: current drug delivery approaches[J/OL]. Respir Res, 2006, 7: 118. DOI: 10.1186/1465-9921-7-118.
[37] Misra A, Hickey AJ, Rossi C, et al. Inhaled drug therapy for treatment of tuberculosis[J]. Tuberculosis, 2011, 91(1): 71-81.
[38] Hirota K, Tomoda K, Inagawa H, et al. Stimulation of phagocytic activity of alveolar macrophages toward artificial microspheres by infection with mycobacteria[J]. Pharm Res, 2008, 25(6): 1420-1430.
[39] Patel B, Gupta N, Ahsan F. Particle engineering to enhance or lessen particle uptake by alveolar macrophages and to influence the therapeutic outcome[J]. Eur J Pharm Biopharm, 2015, 89: 163-174.
[40] Park JH, Jin HE, Kim DD, et al. Chitosan microspheres as an alveolar macrophage delivery system of ofloxacin via pulmonary inhalation[J]. Int J Pharm, 2013, 441(1–2): 562-569.
[41] Maretti E, Rossi T, Bondi M, et al. Inhaled solid lipid microparticles to target alveolar macrophages for tuberculosis[J]. Int J Pharm, 2013, 462(1-2): 74-82.
[42] Garcia Contreras L, Sung J, Ibrahim M, et al. Pharmacokinetics of inhaled rifampicin porous particles for tuberculosis treatment: insight into rifampicin absorption from the lungs of guinea pigs[J]. Mol Pharm, 2015, 12(8): 2642-2650.
[43] Chuan J, Li Y, Yang L, et al. Enhanced rifampicin delivery to alveolar macrophages by solid lipid nanoparticles[J]. J Nanopart Res, 2013, 15(5): 1-9.
[44] Gaspar DP, Faria V, Gon?alves L, et al. Rifabutin-loaded solid lipid nanoparticles for inhaled antitubercular therapy: Physicochemical and in vitro, studies[J]. Int J Pharm, 2015, 497(1-2): 199-209.
[45] Eedara BB, Tucker IG, Das SC. Phospholipid-based pyrazinamide spray-dried inhalable powders for treating tuberculosis[J]. Int J Pharm, 2016, 506(1-2):174-183.
[46] Parumasivam T, Chan JG, Pang A, et al. In vitro evaluation of novel inhalable dry powders consisting of thioridazine and rifapentine for rapid tuberculosis treatment[J]. Eur J Pharm Biopharm, 2016, 107: 205-214.
[47] Goyal AK, Garg T, Rath G, et al. Development and characterization of nanoembedded microparticles for pulmonary delivery of antitubercular drugs against experimental tuberculosis[J]. Mol Pharm, 2015, 12(11): 3839-3850.
[48] Garg T, Goyal AK, Rath G, et al. Spray-dried particles as pulmonary delivery system of anti-tubercular drugs: design, optimization, in vitro and in vivo evaluation[J/OL]. Pharm Dev Technol, 2015, 1-10. DOI: 10.3109/10837450.2015.1081613.
[49] Parumasivam T, Leung SS, Quan DH, et al. Rifapentineloaded PLGA microparticles for tuberculosis inhaled therapy: Preparation and in vitro aerosol characterization[J]. Eur J Pharm Sci, 2016, 88: 1-11.
