CdF分子基态和低激发态的多参考组态理论研究
2016-10-12赵书涛丁海宁周理想张志国王丽萍吴言宁张开银
赵书涛,丁海宁,周理想,武 山,张志国,王丽萍,吴言宁,张开银
(阜阳师范学院 物理与电子工程学院,安徽 阜阳 236037)
CdF分子基态和低激发态的多参考组态理论研究
赵书涛,丁海宁,周理想,武山,张志国,王丽萍,吴言宁,张开银
(阜阳师范学院 物理与电子工程学院,安徽 阜阳 236037)
采用含Davidson修正的多参考组态相互作用方法(MRCI+Q),对F原子和Cd原子分别使用相关一致基augcc-pVQZ和相对论赝势基aug-cc-pwCVQZ-PP,对CdF分子对应最低两个离解限Cd(1Sg)+F(2Pu)和Cd(3Pu)+F(2Pu)的14个电子态2Σ+(3),2Π(3),4Σ+(2),4Π(2),2,4Δ,2,4Σ-,在核间距R=1.3-6.0Å之间进行了Λ-S态势能曲线计算。理论计算结果表明,X2Σ+,22Σ+,22Π,32Σ+为束缚态,而12Π,32Π,12Σ-,12Δ,14Σ+,24Σ+,14Π,24Π,14Δ,14Σ-为排斥态,两个离解限能量间隔为30 147 cm-1。随后,依据Λ-S束缚态势能曲线,对X2Σ+,22Σ+和22Π进行了振动能级和转动常数的计算。上述计算结果跟已有的实验值比较接近。
CdF;势能曲线;振动能级;转动常数
Cd为IIB族元素,它的氟化物具有显著的生物毒性[1-3],伴随着现代工业研究的不断发展,含有这类氟化物的废弃物通过各种渠道排放到人类的生活环境中,对人们的身体造成极大危害。检测这类氟化物的排放需要获得它们的精确光谱数据,然而,由于实验技术和理论方法的制约,相关的光谱数据非常少。
Zn为Cd的同族元素,它的氟化物自由基的实验和理论方面的研究较多。实验方面,Moravec 和Flory等人获得了ZnF自由基的X2Σ+态的结构和转动分辨的光谱常数[4,5]。Rochester和Olsson给出了 A2Π1/2和 A2Π3/2态的绝热激发能分别为4.586 eV和4.632 eV[6]。理论方面,Hayashi等人应用多参考组态相互作用(MRCI)方法结合相对论有效芯势(RECP)计算了ZnF自由基最低的两个离解限对应的全部Λ-S态的势能曲线,从理论上模拟出了较低几个束缚态的振动分辨的吸收谱[7]。Leonard等人采用纳入自旋-轨道耦合效应的MRCI方法计算了最低的两个离解限对应的Λ-S态和Ω态的电子结构,讨论了电子态之间的自旋-轨道耦合作用导致的A2Π态的预解离现象[8]。与含有Zn原子的氟化物自由基相比,近些年关于含有Cd原子的氟化物自由基的实验和理论研究相对较少,赫兹堡等人[9]总结了早期研究给出Cd的氟化物自由基的电子态光谱数据。由于受到早期实验条件的制约,这些自由基的基态和激发态的光谱数据均不完备。近年来,关于含Cd原子的氟化物自由基的理论数据非常少,目前还没有相关的理论文献。不过,作为Cd的最简单化合物,它的氢化物的研究较多[10-12],对较低离解限的Λ-S和Ω态进行了计算,表明对含Cd原子的自由基的理论研究正在兴起。本文对Cd的卤化物自由基CdF的基态和较低激发态进行高精度的从头算,计算中未考虑自旋-轨道耦合效应,只对最低两个离解通道的Λ-S态进行计算。
采用MRCI方法和相关一致基aug-cc-pVQZ (F原子)[13]及相对论赝势基aug-cc-pwCVQZ-PP (Cd原子)[14],在Molpro程序下计算CdF分子对应于最低两个解离通道的Λ-S态势能曲线。利用这些势能曲线的计算结果,借助LEVEL程序求解Schrödinger方程,得到束缚Λ-S态的光谱参数。
1 计算方法
对于较大的双原子分子CdF,要得到它的精确的势能曲线就必须采用多参考组态相互作用(MRCI)理论进行准确的相关能计算。本文采用MRCI方法在MOLPRO2010[15]程序下进行了势能曲线的精确计算。计算过程中,F原子采用了相关一致基aug-cc-pVQZ,对于电子数较多的Cd原子,出于目前实验室计算硬件的限制,则使用了相对 论 赝 势 基 组 aug-cc-pwCVQZ-PP(Cd:ECP28MDF)。
在势能曲线的计算中,MRCI方法只能使用Abelian点群。CdF分子的对称性为C∞v群,然而,由于程序的局限,计算中必须由C2v群代替。C2v群的不可约表示为A1,B1,B2,A2,和C∞v群有以下对应关系:Σ+=A1,Π=B1+B2,Δ=A1+A2和Σ-=A2。在1.3~6.0Å之间给定的核间距上,采用Hartree-Fock(HF)自洽场方法获得了CdF分子基态的分子轨道,以此为初始波函数,采用完全活性空间自洽场(CASSCF)方法对波函数进一步优化。我们选取F原子的2p和Cd原子的5s5p价壳层对应的分子轨道作为CASSCF计算的活性空间轨道。随后,以CASSCF波函数为参考,考虑Davidson修正,利用MRCI方法计算了14个Λ-S态的本征能量。在MRCI计算中,由于Cd原子采用相对论赝 势 基aug-cc-pwCVQZ-PP,冻 结 了1s22s22p63s23p63d10电子,故F原子的1s2和Cd原子的4s24p6电子被放入内核轨道,而F原子的2s2和Cd原子的4d10电子作为关联电子。这样,F原子的2s2和Cd原子的4d10电子的内壳层-价壳层电子关联被考虑进来,在MRCI计算中CdF分子的19个电子的关联效应被考虑进来。
最后,在Λ-S态势能曲线的计算结果上,利用LEVEL[16]程序数值积分来求解CdF分子的一维核运动Schrödinger方程,得到相关束缚态的振动能级和转动常数。
2 研究结果
2.1离解限和电子态
F原子的基电子组态为1s22s22p5,对应的谱项为2Pu,Cd原子的最低两个电子态为[Kr]4d105s2和[Kr]4d105s5p,对应的谱项为1Sg和3Pu,1Pu。由美国国家标准与技术研究院(NIST)的数据可知对应CdF分子的最低2个离解限为Cd(1Sg)+F(2Pu)和Cd(3Pu)+F(2Pu)。Cd(1Pu)+F(2Pu)是CdF分子更高的离解通道,考虑此通道对应的分子态,计算量将增大。鉴于目前的计算条件,本文只涉及最低的两个离解通道对应分子态的理论计算。
F、Cd原子的原子态和CdF分子的分子态所属点群分别为SO(3)和C∞v,它们的不可约表示对应关系为Sg→Σ+,Pu→Σ++Π。当Cd(1Sg)原子和F (2Pu)原子形成CdF分子时对称性降低,SO(3)群的不可约表示分解为C∞v的不可约表示的直和,再通过直积得到对应分子的可能的电子态。通过式(1),可以得到第一离解限Cd(1Sg)+F(2Pu)对应的电子态为2Σ+和2Π。进一步通过式(2),可以得到第二离解限 Cd(3Pu)+F(2Pu)对应的电子态为2,4Σ+(2),2,4Σ-,2,4Π(2)和2,4Δ。

对CdF分子电子态的MRCI势能曲线的计算就按照上述两个离解极限进行,其中涉及二重态的有8个:2Σ+(3),2Π(3),2Σ-,2Δ;四重态的有6个:4Σ+(2),4Π(2),4Σ-,4Δ。Λ-S态与离解限的关联表及计算结果见表1。

表1 Λ-S态与解离限关联表
从表1中可以看出,14个Λ-S态对应的两个离解限间距的计算结果30 147 cm-1跟NIST给出的实验值[17]31 246 cm-1比较接近,偏差为1 099 cm-1,这为本文后续的理论计算提供可靠的基础。
2.2势能曲线
在MRCI+Q方法下对Cd(1Sg)+F(2Pu)和Cd (3Pu)+F(2Pu)两个离解限对应的14个Λ-S态进行了势能曲线的扫描计算,结果如图1和2所示,其中图1为二重态势能曲线,图2为四重态势能曲线。
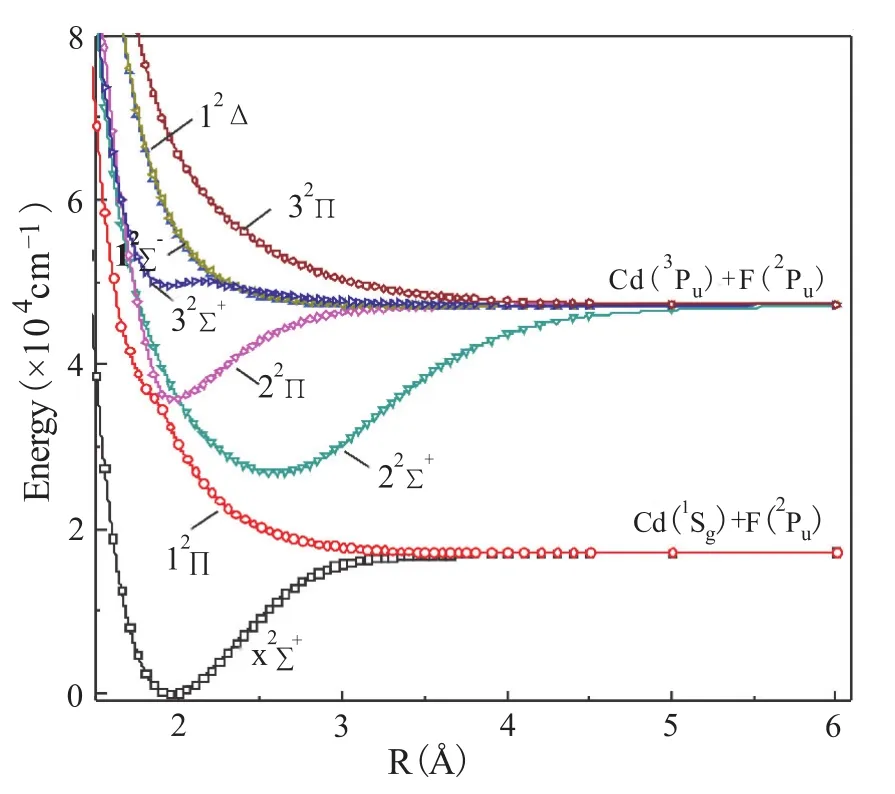
图1 MRCI+Q计算的CdF分子的二重态势能曲线
从图中可以看出,X2Σ+,22Σ+,22Π,32Σ+为束缚态,而12Π,32Π,12Σ-,12Δ,14Σ+,24Σ+,14Π,24Π,14Δ,14Σ-为排斥态。基态X2Σ+的平衡位置在R=1.95 Å附近,在核间距R=2.60 Å处与相同对称性的激发态22Σ+有强烈的相互作用。22Σ+和32Σ+对应于离解限Cd(3Pu)+F(2Pu),由于相同对称性的避免交叉,22Σ+态有一个深势阱,在R=2.60 Å处有极小值,32Σ+有一个很浅的势阱,只能容纳1个振动能级,在R=1.95 Å处有极小值。由于相同的对称性,12Π和22Π在R=1.90 Å附近发生避免交叉,导致两态分别对应于离解限Cd(1Sg)+F(2Pu)和Cd(3Pu)+F (2Pu)。由于避免交叉现象,22Π有一个较深的势阱,在R=1.95 Å处有极小值。
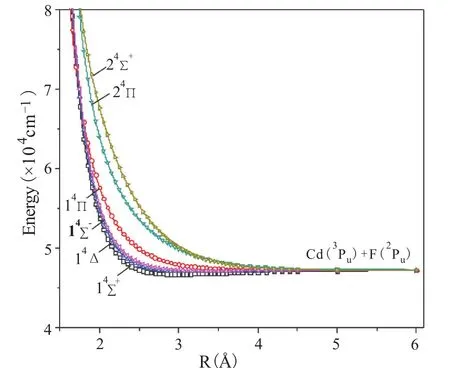
图2 MRCI+Q计算的CdF分子四重态势能曲线
在基态X2Σ+的平衡位置R=1.95 Å处给出了较低的Λ-S态X2Σ+,12Π,22Π,22Σ+,32Σ+和12Σ-的垂直激发能和主要组态。12Π,22Π和22Σ+的分布在32 000~38 000 cm-1区间,相互作用较强。12Π,22Π,22Σ+,32Σ+和 12Σ-的主要组态分别为6σ27σ28σ03π34π0,6σ27σ08σ03π44π1,6σ17σ28σ03π44π0,6σ27σ08σ13π44π0和 6σ27σ18σ03π34π1分别对应于3π→7σ,7σ→4π,6σ→7σ,7σ→8σ和3π→4π单电子跃迁。
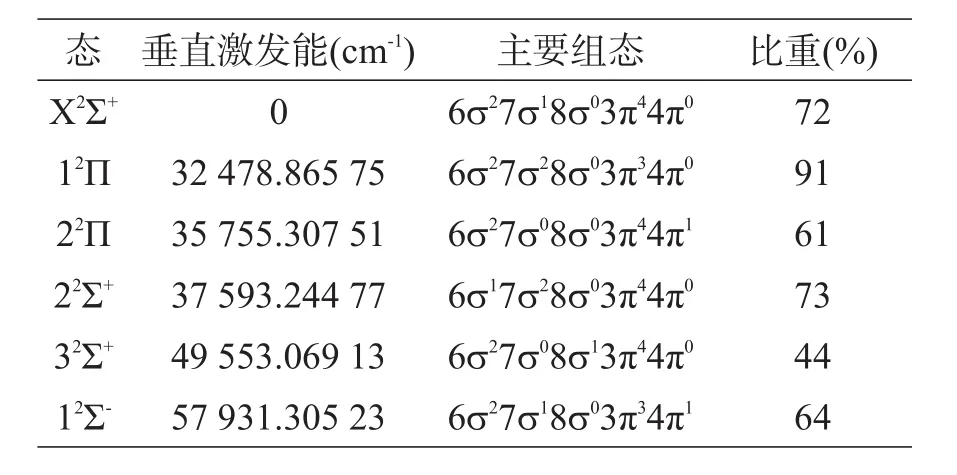
表2 较低Λ-S态的垂直激发能和主要组态R=1.95 Å
2.3振动能级和转动常数
在Λ-S态势能曲线基础上,通过LEVEL程序求解CdF核运动的薛定谔方程,得到了X2Σ+,22Σ+和22Π的振动能级和转动常数。对应上述3个束缚态分别找到49,91和32个振动能级,鉴于篇幅限制,只给出了前32个振动能级。

表3 X2Σ+,22Σ+和22Π的振动能级G(v)和转动常数Bv
3 结论
采用MRCI+Q方法对CdF分子对应于Cd (1Sg)+F(2Pu)和Cd(3Pu)+F(2Pu)离解限的14个电子态2Σ+(3),2Π(3),4Σ+(2),4Π(2),2,4Δ,2,4Σ-,进行了高精度的从头计算。两个离解限能量间隔为30 147 cm-1与实验结果符合良好,X2Σ+,22Σ+,22Π,32Σ+为束缚态,而其它10个态(12Π,32Π,12Σ-,12Δ,14Σ+, 24Σ+,14Π,24Π,14Δ,14Σ-)均为排斥态,预测的X2Σ+,22Σ+和22Π的振动能级和转动常数可为后续的实验和理论研究提供重要参考。
[1] Koren Š,Arčon I,Kump P,et al.Influence of CdCl2 and CdSO4 supplementation on Cd distribution and li-gand environment in leaves of the Cd hyperaccumulator Noccaea(Thlaspi)praecox[J].Plant and Soil,2013,370(1/2):125-148.
[2] Meyer C L,Juraniec M,Huguet S A,et al.Intraspecific variability of Cadmium tolerance and accumulation,and cadmium-induced cell wall modifications in the metal hyperaccumulator Arabidopsis halleri[J].Journal of Experimental Botany,2015,66(11):3215-3227.
[3]Isaure M P,Huguet S,Meyer C,et al.Evidence of various mechanisms of Cd sequestration in the hyperaccumulator Arabidopsis halleri,the non-accumulator Arabidopsis lyrata,and their progenies by combined synchrotron-based techniques[J].Journal of Experimental Botany,2015,66(11):3201-3214.
[4] Moravec V D,Klopcic S A,Chatterjee B,et al.The electronic structure of ZnO and ZnF determined by anion photoelectron spectroscopy[J].Chemical Physics Letters,2001,341(3/4):313-318.
[5] Flory M A,Mclamarrah S K,Ziurys L M.Completing the 3d metal fluoride series:The pure rotational spectrum of ZnF(X-2 Sigma(+))[J].Journal of Chemical Physics,2006,125(19):194304-194309.
[6] Rochester G,Olsson E.Das bandenspektrum des MnF [J].Zeitschrift für Physik,1939,114(7/8):495-499.
[7] Hayashi S,Leonard C,Chambaud G.Ab initio study of the low lying electronic states of ZnF and ZnF-[J]. The Journal of Chemical Physics,2008,129(4):044313.
[8]Leonard C,Le Quere F.Theoretical study of the predissociation of the A(2)Pi state of ZnF including quasi-diabatisation of the spin-orbit coupling[J].Journal of Chemical Physics,2012,137(16):164318.
[9]Huber K,Herzberg G.IV of molecular spectra and molecular structure[M].New York:Van Nostrand Reinhold,1979.
[10]Li Rui,Zhai Zhen,Zhang Xiaomei,et al.Spin-orbit all-electron configuration interaction study on the electronic structure and radiative lifetimes of low-lying excited states of CdH[J].Chemical Physics Letters,2014,599:51-56.
[11]Zhang Xiaomei,Liang Guiying,Li Rui,et al.Multireference configuration interaction study on the potential energy curves and radiative lifetimes of low-lying excited states of CdH+cation[J].Chemical Physics,2014,443:142-148.
[12]Abe M,Kajita M,Hada M,et al.Ab initio study on vibrational dipole moments of XH+molecular ions:X= 24Mg,40Ca,64Zn,88Sr,114Cd,138Ba,174Yband 202Hg[J].Journal of Physics B-Atomic Molecular and Optical Physics,2010,43(24):245102-245112.
[13]Dunning J H.Gaussian basis sets for use in correlated molecular calculations.I.The atoms Boron through Neon and Hydrogen[J].The Journal of Chemical Physics,1989,90(2):1007-1023.
[14]Peterson K A,Puzzarini C.Systematically convergent basis sets for transition metals.II.Pseudopotentialbased correlation consistent basis sets for the group 11 (Cu,Ag,Au)and 12(Zn,Cd,Hg)elements[J].Theoretical ChemistryAccounts,2005,114(4/5):283-296.
[15]Werner H J,Knowles P J,Lindh R,et al.MOLPRO,A package of ab initio programs[Z],2010.
[16]Le Roy R J.LEVEL 8.0 A computer program for solving the radial Schrödinger equation for bound and quasibound levels,University of Waterloo:Chemical Physics Research Report NO.CP-663,2007.
[17] Burns K,Adams K B.Energy levels and wavelengths of natural cadmium and of cadmium-114[J].Journal of the Optical Society ofAmerica,1956,46(2):94-99.
MRCI calculations for ground and excited states of CdF molecule
ZHAO Shu-tao,DING Hai-ning,ZHOU Li-xiang,WU Shan,ZHANG Zhi-guo,WANG Liping,WU Yan-ning,ZHANG Kai-yin
(School of Physics and Electronic Engineering,Fuyang Normal University,Fuyang Anhui 236037,China)
The potential energy curves(PECs)of CdF correlating to the first two dissociation asymptotes Cd(1Sg)+F(2Pu)and Cd(3Pu)+F(2Pu)were computed by using multi-reference configuration interaction including Davidson correction(MRCI+Q)approach.The PECs of the fourteen states(2Σ+(3),2Π(3),4Σ+(2),4Π(2),2,4Δ,and2,4Σ-)were calculated in the internuclear distance range from 1.3 to 6.0 angstrom at the basis sets,aug-cc-pVQZ for F and aug-cc-pwCVQZ-PP for Cd atom.The calculated results show that X2Σ+,22Σ+,22Π,32Σ+are bound,while 12Π,32Π,12Σ-,12Δ,14Σ+,24Σ+,14Π,24Π,14Δ,14Σ-are repulsive.The energy gap of the second and the first dissociation limit is 30 147 cm-1.Based on the computed PECs of X2Σ+,22Σ+and 22Π,the vibrational level and rotational constant were obtained,which are in good accordance with the available experimental data.
CdF;potential energy curves;vibrational levels;rotational constants
O561
A
1004-4329(2016)02-028-05
10.14096/j.cnki.cn34-1069/n/1004-4329(2016)02-028-05
2016-02-10
安徽省教育厅项目(2014KJ020);安徽自然科学基金项目(1608085MA10);安徽省大学生创新训练项目(AH201410371076);阜阳师范学院科研项目(2014FSKJ04ZD)资助。
赵书涛(1982-),男,博士,讲师,研究方向:原子与分子物理、光学。
