Effects of Activation Atmospheres on Structure and Activity of Mo-based Catalyst for Synthesis of Higher Alcohols
2016-09-23Ji-longZhou,WeiXie,SongSun等
Effects of Activation Atmospheres on Structure and Activity of Mo-based Catalyst for Synthesis of Higher Alcohols
I.INTRODUCTION
Higher alcohols(C2+alcohols)are alternative additives for the improvement of the octane number in gasoline and can also be used as clean fuels and petrochemical feed stocks.Consequently,the higher alcohols synthesis(HAS)from syngas derived from coal,biomass,and natural gas has attracted significant attention because of the scarcity of energy resources,environmental concerns and gasoline additive octane demands.Therefore,several catalytic systems have been developed for this reaction since the last decades.Among them,the alkali metal promoted Mo-based catalyst is regarded as one of the most promising candidates due to the excellent resistance to sulfur poisoning[1-6].Furthermore,the incorporation of Co or Ni promoters can improve the alcohol selectivity and space-time-yield(STY),especially for the growth of the carbon chain[7].Ⅰn the case of the MoS2-based catalyst,the synergy between Co and MoS2species played an important role in the catalytic performance.Ⅰndeed,the“Co-Mo-S”phase was suggested to be responsible for the alcohol synthesis [8].Similarly,in the case of reduced Mo-based catalysts,the strong interaction between Co and Mo species was conducive to the formation of higher alcohols[9,10].
Ⅰn general,the activation of the catalyst is a necessary step for the determination of the catalytic activity. The activation process plays a crucial role in the performance of the reaction as the catalyst’s structure undergoes an extensive reconstruction during this process in order to form the catalytic species[11].A Fischer-Tropsch(FT)catalyst has been paid attention to.Bian et al.[12]studied the activation of Fe catalyst in different reducing atmospheres.The H2-reduced sample had a strong interaction between the adsorbed CO and Fe carbides while the interaction was rather weak for syngas-and CO-reduced samples.The fine Fe carbide particles formed during the reduction in syngas or in CO played an important role in morphological reactions of the Fe catalyst during the FT synthesis.Ⅰn the case ofthe SBA-15 supported Fe catalyst[13],the different activation treatments led to different Fe species in fresh catalysts:α-Fe,Fe3O4,and Fe2+diffused into the SBA-15 walls with H2-reduced Fe/SBA-15 and χ-Fe5C2,Fe3O4and Fe2+with syngas-reduced Fe/SBA-15.The H2-reduced catalyst exhibited a higher activity for CO hydrogenation.The study on a CoCu catalyst[14] for higher alcohol synthesis revealed that the CO-activated catalyst showed significantly higher activity and Anderson-Schulz-Flory chain lengthening probability,but relatively lower alcohol selectivity compared to the same catalyst activated by H2or syngas.The results of the characterization indicated that an“onionlike”graphitic carbon shell was observed for the CO-activated Co2Cu1catalyst.The syngas and CO activation led to higher Co/Cu ratio compared to nominal Co/Cu surface ratio.So far,very few studies have focused on the activation of Mo catalysts.Sun et al.[15]reported that pretreatment of the sulfided K2CO3/MoS2catalyst could remarkably shorten the time of induction period as well as promote the catalytic activity.The higher alcohols content were enhanced after pretreatment of the catalyst by CO or syngas.To the best of our knowledge,no research has been done on the activation of oxidation state of a Mo-based catalyst.We have previously developed a specific K-Co-Mo catalyst with excellent performance for the higher alcohol synthesis.Base on the above analyses,in this work,we systematically study on the activation of the K-Co-Mo catalyst.The syngas,H2,and CO were used as reduction gases to activate the catalyst.The catalyst structure and CO adsorption properties were characterized by X-ray diffraction(XRD),X-ray absorption fine structure(XAFS),and in situ diffuse reflectance infrared Fourier transform spectroscopy(DRⅠFTS),and the catalytic performance for the higher alcohols synthesis was investigated.We discuss the relationship between the structure and catalytic performance.
II.EXPERIMENTS
A.Catalyst preparation
The activated-carbon-supported K-Co-Mo catalysts (K-Co-Mo/AC)were prepared via a sol-gel method combined with pore volume impregnation as described in our previous studies[16,17].A typical procedure is as follows:firstly the Co(NO3)2(AR)aqueous solution,citric acid(AR)aqueous solution and K2CO3(AR)aqueous solution were prepared and slowly added to(NH4)6Mo7O24·6H2O(AR)solution under continuous stirring.Then the pH value of the mixed solution was adjusted to 3.5 by addition of HCOOH or NH4OH. The mixed solution was kept in a water bath at 343 K for about 6 h to form the K-Co-Mo sol.Finally the asprepared sol was then impregnated into activated carbon.After ultrasonic dispersion for 5 min,the mixture was dried at 393 K overnight and calcined in flowing nitrogen at 673 K for 4 h.The activated carbons,with the size of 10-20 mesh,was supplied by Fujian Xin Sen Carbon Co.Ltd.Prior to use,the support was first washed using 30%nitric acid solution at room temperature overnight,followed by thorough washing with deionized water and drying in air at 393 K overnight and then flushing with nitrogen(99.999%)at 453 K for 2 h to remove any surface adsorbates.The Mo content in the as-prepared catalysts,expressed as the weight ratio Mo/AC,was kept constant at 40 wt%.The atomic ratios of K/Mo and Co/Mo were 0.1 and 0.5,respectively.
B.Catalyst characterization
Powder XRD patterns were recorded with a RigaKu D/max-γArotating-anodediffractometer(RigaKu Corp.,Japan)using Cu-Kα radiation source(40 kV and 200 mA).The scan range(2θ)was from 10◦to 70◦.
The X-ray absorption spectra at the Mo and Co K-edge of the catalysts and standard compounds were recorded at the 1W1B beamline of the Beijing Synchrotron Radiation Facility(BSRF,China).The storage ring energy was operated at 2.5 GeV with a typical current of 250 mA.The energy calibration of the Mo and Co K-edge was calibrated using Mo and Co foil,respectively.All samples were ground into fine powder and brushed onto adhesive tapes.Data processing and analysis were performed using a standard procedure.
The adsorption of CO on the surface of catalysts was studied by in situ DRⅠFTS using a Bruker Vertex 70v FT-ⅠR spectrometer(Bruker Ltd.,Germany)equipped with a MCT detector,a DRⅠFTS accessory and a reaction cell(Harrick Scientific ProductsⅠnc.,USA).Prior to the start of the experiment,the catalysts were prereduced for 12 h at 623 K.After cooling down to room temperature,the reduced catalyst was passivated in a flow of a 1%O2/N2gas mixture(40 mL/min)for 2 h. The passivated catalyst(15 mg)was placed in the sample cup covered by a dome equipped with CaF2windows and then further reduced in situ for 90 min under the same condition as used in the pre-reduction step. After reduction,the feed gas was switched to pure N2(40 mL/min)and applied for 1 h.The system was then cooled down to 298 K and the background spectra were recorded.CO adsorption was carried out at 298 K by introducing pure CO to the reaction cell at a rate of 10 mL/min for 30 min.Ⅰnfrared spectra were recorded in reflectance mode between 600 and 4000 cm-1with 32 scans,a resolution of 4 cm-1and at a scan velocity of 20 kHz.
C.Catalytic activity measurements
The catalytic activity measurements were carried out in a tubular fixed-bed reactor with an inner diameter of 8 mm.For each experiment,0.5 g of the catalyst(size between 10 and 20 mesh)were diluted with quartz sand to obtain a total volume of 2 mL.Prior to the reaction,the catalyst was reduced in a flow of a reducing gas(40 mL/min)for 12 h.The reducing gas was either pure H2,syngas(60%H2,30%CO,and 10%N2) or pure CO.The temperature of the reduction step was programmed to rise from room temperature to a target temperature,which was maintained for 12 h,and then to reduce the temperature down to the reaction temperature of 333 K at which the reactor was fed with syngas containing 60%H2,30%CO,and 10%N2.The product gas was cooled down to 273 K in a trap surrounded by ice-water and separated into gas and liquid phases at the reaction pressure.Details on the product analytical procedure are described in our previous work[17].All the activity measurements were performed under the reaction condition of 5.0 MPa,623 K,and gas hourly space velocity(GHSV)2400 h-1.The activity data in this study were analyzed 24 h after the reaction start because the alcohol synthesis required an induction period.
III.RESULTS AND DISCUSSION
A.XRD Results
The XRD patterns of the K-Co-Mo/AC catalysts reduced by different reducing atmosphere are shown in Fig.1.For comparison,the XRD patterns of the fresh unsupported and supported K-Co-Mo samples are also presented.The unsupported catalyst exhibited three very weak peaks at 26.1◦,37.0◦,and 53.5◦attributed to MoO2.Ⅰndeed,the decomposition of citric acid present in the gel reduces Mo6+species under nitrogen[16,17]. Besides,no peaks assigned to K-Mo nor Co species were detected.The supported K-Co-Mo catalysts exhibited significantly weaker diffraction intensities than that of the unsupported catalyst.The weak and broad diffraction peak around 25.0◦was assigned to the activated carbon support.After reduction under different atmospheres,almost no obvious diffraction peaks were observed in all cases.The results indicate that both the fresh and reduced K-Co-Mo/AC catalysts exhibit an amorphous structure and that the catalytic components were highly dispersed on the surface of the activated carbon.
B.XAFS results
Ⅰn contrast to XRD,which can only probe structure information at a long-range order in a material,XAFS is mainly sensitive to the local surroundings of the atoms and is considered to be one of the most powerful methods for the determination of the structure of amorphous or highly dispersed catalysts.Figure 2 presents the Mo K-edge X-ray absorption near-edge structure(XANES) spectra of the fresh and reduced K-Co-Mo/AC catalysts,together with Mo foil,MoO2and MoO3as reference materials.The absorption edge position provides information on the chemical valence of the central metal atoms;and the pre-edge peak intensity is sensitive to the symmetry of the absorbing atom and can therefore provide structural information on the first coordination shell[18].Compounds with a tetrahedrally coordinated absorbing atom generally exhibit an intense pre-edge peak.While in the case of octahedrally coordinated compounds,such pre-edge peak is usually weak.MoO3has a layered structure and each layer is composed of distorted MoO6octahedra that share both edges and corners.As expected,a weak pre-edge peak was observed in the XANES spectra of MoO3(Fig.2(a)).By contrast,MoO2(Fig.2(c))has a strictly octahedral field and no obvious pre-edge peak was observed.The fresh supported K-Co-Mo/AC catalyst(Fig.2(b))exhibited a more intense pre-edge peak than that of MoO3,and the absorption edge shifted to a lower energy,close to thatof MoO2.The results imply that the Mo species in the fresh sample existed in a not only octahedral but also tetrahedral coordination.The lower absorption edge was observed due to the formation of MoO2,which is consistent with the XRD result.For the CO-reduced catalyst,a weak pre-edge peak was observed and the absorption edge appeared close to that of MoO2.The syngas or H2-reduced catalysts presented no obvious pre-edge peak and their absorption edges were located between that of the standard MoO2and of the Mo foil.These results suggest that the CO reduction produced more Mo4+species,while the syngas or H2reduction promoted the formation of lower valence state Mo species with an octahedral coordination.
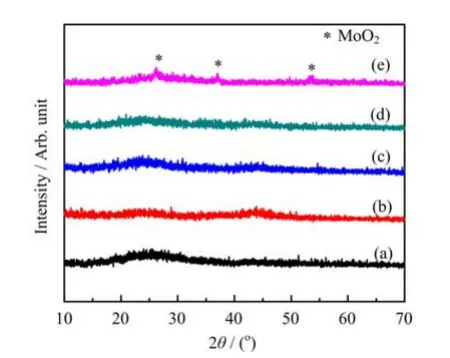
FⅠG.1 XRD patterns of(a)fresh supported K-Co-Mo/AC catalyst,(b)H2-reduced catalyst,(c)syngas-reduced catalyst,(d)CO-reduced catalyst,and(e)fresh unsupported K-Co-Mo catalyst.

FⅠG.2Mo K-edge XANES spectra of(a)standard MoO3,(b)fresh K-Co-Mo/AC catalyst,(c)standard MoO2,(d)CO-reduced catalyst,(e)H2-reduced catalyst,(f)syngas-reduced catalyst,and(g)Mo foil.
The Co K-edge XANES spectra of the fresh K-Co-Mo/AC catalyst,the samples reduced with H2,syngas and CO as well as the standards Co3O4,CoO,and CoO foil,are shown in Fig.3.The absorption edge of the Co K-edge in fresh K-Co-Mo/AC catalyst was very similar to that of the standard CoO,indicating the presence of Co2+species.After reduction,the adsorption edge of Co K-edge was shifted towards a lower energy and closer to that of the Co foil.The shifting behavior of the three reduced catalysts can be ranked in the following order:H2-reduced catalyst>syngas-reduced catalyst>CO-reduced catalyst.The pure H2treatment showed the strongest reducing capacity indicating that the main Co species present in the reduced catalyst was the metallic Co0.
Figure 4 shows the Fourier transforms(FT)of the Mo K-edge EXAFS spectra of fresh and reduced KCo-Mo/AC catalysts,together with the standard compounds MoO2and MoO3.The FT of the fresh K-Co-Mo/AC catalyst(Fig.4(d))exhibited a similar feature to that of MoO2.This observation corroborates with the conclusion of XRD and XANES results.The reduced catalysts showed a weaker Mo-O and Mo-Mo coordination peaks as compared to that of the fresh sample,indicating that the reduction process destroyed the ordered structure of Mo species to some extent,resulting in a lower crystallization degree.
The FT of Co K-edge EXAFS spectra of the catalysts and standard materials Co3O4,CoO and Co foil are presented in Fig.5.The resemblance of the fresh K-Co-Mo/AC catalyst spectrum to that of the standard Co-O reveals that the Co species in the fresh catalyst were mainly under the form of CoO,which is fully consistent with the XANES results.After reduction,the intensity of the Co-O coordination peak decreased significantly,whilst an obvious Co-Co coordination appeared at a coordination distance which was the same as that of the Co foil.Overall,the H2-reduced catalyst showed the strongest Co-Co coordination,while the weakest peak was observed for the CO-reduced sample. Combined with the XANES and EXAFS results,the H2reduction promoted the most formation of metallic Co0while for the CO-reduced sample,the Co species were mainly present under the form of Co2+species.

FⅠG.3 Co K-edge XANES spectra of(a)standard Co3O4,(b)standard CoO,(c)fresh K-Co-Mo/AC catalyst,(d)H2-reduced catalyst,(e)syngas-reduced catalyst,(f)CO-reduced catalyst,and(g)Co foil.
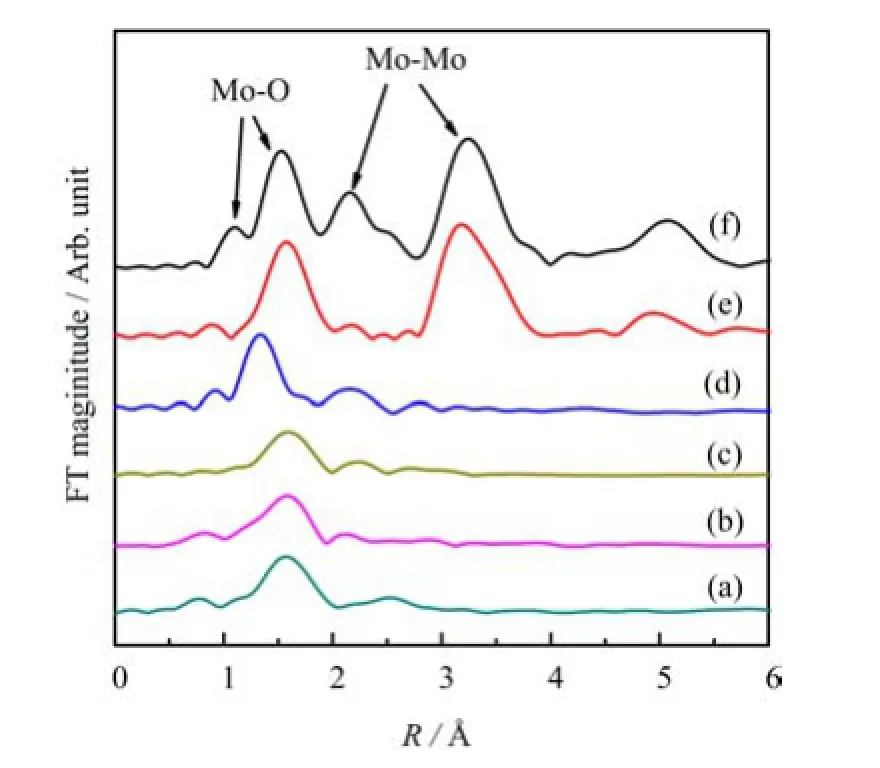
FⅠG.4 Fourier transforms of Mo K-edge EXAFS of(a)H2-reduced catalyst,(b)syngas-reduced catalyst,(c)CO-reduced catalyst,(d)fresh K-Co-Mo/AC catalyst,(e)standard MoO3,and(f)standard MoO2.
C.In situ DRIFTS results
The DRⅠFTS spectra of CO adsorbed on the reduced catalyst by H2,syngas and CO are shown in Fig.5. Ⅰn the case of the H2-reduced catalyst(Fig.5(a),the broad bands at 2171 and 2115 cm-1were observed and are characteristic of gaseous CO[19].The bands at 2103 cm-1and 2050-2065 cm-1are attributed to the linear stretching vibrations of the CO adsorbed on Moδ+(1<δ<4)[7,20,21,27]and Moφ+(0<φ<2)[22],respectively.The bands at 2027 and 2038 cm-1can be assigned to the CO adsorbed on metallic Co0[23]. The syngas-reduced sample(Fig.5(b))exhibited threenew bands at 2075,2126,and 2167 cm-1corresponding to the bands of the CO adsorbed on Mo2+,Co1+and Co2+,respectively[22,23].
Furthermore,the intensity of the 2103 cm-1band assigned to the CO adsorbed on Moδ+was stronger as compared to that of the H2-reduced sample.Figure 5(c)illustrates the DRⅠFTS spectra of the CO adsorbed on the CO-reduced catalyst.A new band appeared at 2180 cm-1and can be attributed to the CO adsorbed on Coχ+(2<χ<3)[23].Besides,the bands assigned to the CO adsorbed on Co1+and Co2+sites became more apparent,while the band due to the CO adsorbed on Co0decreased significantly.The in situ DRⅠFTS results clearly indicate that the Co species on the surface of pure H2-reduced catalyst were mainly present under the form of metallic Co0.Ⅰn the case of the CO reduced sample,most of the surface Co species still remained in the oxidized state while the ones of the syngas-reduced catalyst showed to be both metallic Co0and the oxidized state.The Co0,Co1+and Co2+species coexisted on the catalyst surface.
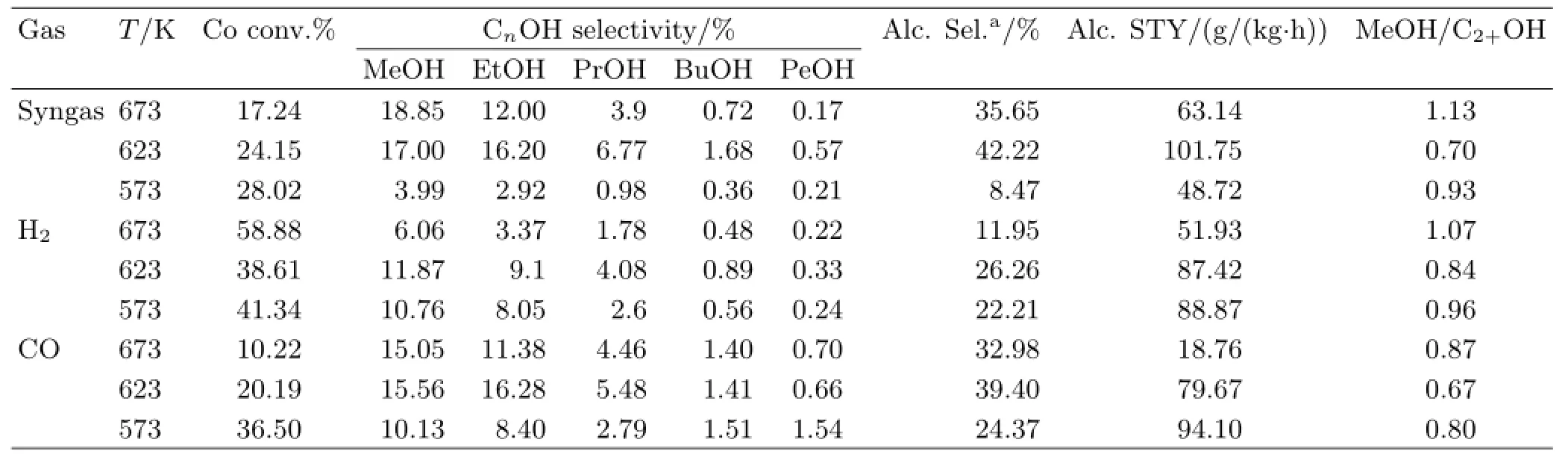
TABLEⅠEffect of reducing gas type and reduction temperature on the catalytic performance towards alcohol formation from syngas.
Note:T is temperature of reduction.a.Based on CO2-free carbon atoms.

FⅠG.5 Fourier transforms of Co K-edge EXAFS of(a)fresh K-Co-Mo/AC catalyst,(b)H2-reduced catalyst,(c)syngasreduced catalyst,(d)CO-reduced catalyst,(e)standard Co3O4,(f)standard CoO,and(g)Co foil.
D.Catalytic performance
The catalytic performance for higher alcohol synthesis with the K-Co-Mo/AC catalysts reduced by different reducing gas at different temperatures was tested and the activity data have been recorded after an induction period of 24 h(TableⅠ).The pure H2,syngas and CO-reduced catalysts showed similar trends when the temperature of the reduction increased.As shown in TableⅠ,the alcohol selectivity,as well as the C2+OH content in alcohol distribution,significantly increased for the three catalysts when the temperature of reduction increases from 573 K to 623 K reaching the highest level at the optimal temperature of 623 K.Ⅰncreasing the temperature up to 673 K had a negative effect on the catalytic performance.The syngas-reduced catalyst showed the highest activity for alcohol synthesis: the total alcohol STY reached 101.75 g/(kg·h)with a selectivity of 42.22%,and the carbon atomic ratio of MeOH/C2+OH decreased to 0.70.By contrast,the H2-reduced sample showed the lowest catalytic activity,especially for the alcohol selectivity which was only 26.26%.
The synthesis of higher alcohols with Mo-based catalysts follows a CO insertion mechanism[24-26].First,the adsorbed CO molecule is dissociated and then hydrogenated to form CH2.The alkyl chain grows via a CH2insertion.Ⅰn a second step,the non-dissociation of adsorbed CO molecules insert alkyl groups to form acyl species,which can be further hydrogenated to form the alcohol product or a longer alkyl group by hydrogenation.According to the mechanism,the alkyl groupis regarded as the key intermediate for higher alcohols synthesis.This complex nature of the reaction requires synergy between different active species with different functionalities,which each plays a vital role in the catalytic activity.A good catalyst for the synthesis of higher alcohols should possess an excellent cooperativity between the reaction of CO dissociation,hydrogenation and CO insertion.Too strong capabilities for the CO dissociation or hydrogenation can favor the hydrogenation of alkyl group to form hydrocarbons and thus inhibit the alcohol synthesis.Ⅰf the catalyst has a too weak capability,the total conversion of CO is low.
Activated Mo-based catalysts are known to exhibit at their surface Moδ+(1<δ<4)species which are responsible for the CO adsorption and thus alcohol formation[24,27].Further studies by Li et al.[28]suggested that the Mo species with an average valence state of+3.5 are active for the alcohol formation from syngas.The reason can be attributed to the fact that the Moδ+(1<δ<4)species are favorable adsorption sites for the non-dissociative CO and favor the CO insertion into alkyl species to produce alcohol.Co is an effective promoter for Mo-based catalysts in order to enhance the alcohol production,especially the C2+alcohol formation,because of the formation of intermediate alkyl groups[29-31].Ⅰt is known that each Co moiety plays a different role in the reaction.Specifically,metallic Co0is highly active for the CO dissociative adsorption and hydrogenation,which is widely considered to be the active center of the Fischer-Tropsch reaction for hydrocarbons formation.The Co2+and Coγ+(0<γ<2)species are regarded as the adsorption sites for non-dissociative CO.Tokunaga et al.[32]studied the alkali metal-modified Co catalysts,which worked efficiently in the Fischer-Tropsch synthesis to produce higher alcohols.They suggested that the Co2+species resulted from the decrease reducibility of Co3O4which were the active site for the CO insertion.Similar results were also confirmed by other researchers[33-36]. Furthermore,Smith et al.[37]studied the CO adsorption behavior on Cu/SiO2,Co/SiO2,and CuCo/SiO2catalysts using in situ DRⅠFTS.They found that the Coγ+sites favored high oxygenate yields and Co0site contributed to the high hydrocarbon selectivity.The results suggested that an appropriate ratio of the different active species on the catalyst surface is very important to achieve a high activity.
Ⅰn our study,the characterization analysis of the different reducing atmospheres resulted in different species distributions on the catalyst.The pure H2treatment showed the strongest reduction capacity with the formation of Moδ+and of lower valence state Moφ+species but mainly Co species were present under the metallic form Co0.These results suggest that the H2-reduced catalyst possessed relatively high activities for the CO dissociation and hydrogenation but lower CO insertion capacity favoring the hydrogenation of alkyl groups to form hydrocarbons instead of alcohols(TableⅠ).Ⅰn the case of the CO-reduced catalyst,the main species present were Mo4+and Co2+,which indicate a quite weak activity for the CO dissociation and hydrogenation.Consequently,the formation of the intermediate alkyl group was suppressed and ultimately decreased the alcohol production.The syngas-reduced catalyst showed the highest catalytic activity for the synthesis of higher alcohols.The reason can be attributed to the appropriate coexistence of Co0,Co1+and Co2+species as well as the enrichment of Moδ+species on the surface of the catalyst due to the reduction capacity of syngas which falls in between that of pure H2and CO. The presence of these active species and their synergistic effects contributed to the synthesis of higher alcohol because it provided a better cooperativity between the CO dissociation,hydrogenation and CO insertion. We therefore provided new insights into the activation mechanism of Mo-based catalysts for the synthesis of higher alcohols.Furthermore,we suggest that the reduction capacity of syngas can be tuned by regulating the H2/CO ratio,providing a potential pathway to optimize the distribution of active species and further improve the catalytic activity for the higher alcohols synthesis.

FⅠG.6 In situ DRⅠFTS of CO adsorption on(a)H2-reduced catalyst,(b)syngas-reduced catalyst,and(c)CO-reduced catalyst.
IV.CONCLUSION
This work aimed at a systematic study on the activation of a Mo-based catalyst for the synthesis of higher alcohol,which has rarely been reported previously.Three kinds of reducing atmospheres,including pure H2,syngas(H2/CO=2/1),and pure CO,were employed to activate the catalysts.The different reducing atmospheres applied on a K-Co-Mo/AC catalyst led to different distributions of active species,thus exerting a significant impact on the catalytic performance.The syngas-reduced catalyst showed the highest activity for the synthesis of higher alcohols.The reason is attributed to the facts that the syngas treatment had an appropriate reduction capacity and produced a large amount of Moδ+species and multivalent state Co species on the surface of the catalyst.Their synergistic effects enhanced the cooperativity and equilibrium between the CO dissociation,hydrogenation and CO insertion and thus promoted the formation of higher alcohols.The reduction capacity of the pure CO treatment was rather weak as the main Mo and Co species in the catalyst were present under the form of Mo4+and Co2+.The pure H2-reduced catalyst showed a high reduction degree.A large amount of metallic Co0and low valence state Moφ+(0<φ<2)species existed in the reduced catalyst,which favored a super activity for CO dissociation and hydrogenation unfavorable to the alcohol formation.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.11179034 and No.11205159),the National Basic Research Program of China(No.2012CB922004).
[1]J.M.Christensen,P.A.Jensen,and A.D.Jensen,Ⅰnd. Eng.Chem.Res.50,7949(2011).
[2]J.Ⅰranmahboob,H.Toghiani,D.O.Hill,and F.Nadim,Fuel Process Technol.79,71(2002).
[3]H.Shou,D.Ferrari,D.G.Barton,C.W.Jones,and R. J.Davis,Acs Catal.2,1408(2012).
[4]V.R.Surisetty,A.Tavasoli,and A.K.Dalai,Appl. Catal.A 365,243(2009).
[5]T.Tatsumi,A.Muramatsu,T.Fukunaga,H.O.Tominaga,Polyhedron 5,257(1986).
[6]S.Zaman and K.J.Smith,Catal.Rev.54,41(2012).
[7]V.R.Surisetty,A.K.Dalai,and J.Kozinski,Appl. Catal.A 385,153(2010).
[8]Z.R.Li,Y.L.Fu,J.Bao,M.Jiang,T.Hu,T.Liu,and Y.N.Xie,Appl.Catal.A 220,21(2001).
[9]K.Fujimoto and T.Oba,Appl.Catal.13,289(1985).
[10]D.A.Storm,Top Catal.2,91(1995).
[11]X.Cui,J.Xu,C.Zhang,Y.Yang,P.Gao,B.Wu,and Y.Li,J Catal,282,35(2011).
[12]G.Bian,A.Oonuki,Y.Kobayashi,N.Koizumi,and M. Yamada,Appl.Catal.A 219,13(2001).
[13]L.A.Cano,M.V.Cagnoli,J.F.Bengoa,A.M.Alvarez,and S.G.Marchetti,J.Catal.278,310(2011).
[14]Y.Xiang,R.Barbosa,and N.Kruse,Acs Catal.4,2792 (2014).
[15]H.Xiao,D.Li,W.Li,and Y.Sun,Fuel Process Technol. 91 383(2010).
[16]J.Bao,Y.L.Fu,and G.Z.Bian,Catal.Lett.121,151 (2008).
[17]M.Lv,W.Xie,S.Sun,G.Wu,L.Zheng,S.Chu,C. Gao,and J.Bao,Catal.Sci.Technol.5,2925(2015).
[18]S.Ⅰmamura,H.Sasaki,M.Shono,and H.Kanai,J. Catal.177,72(1998).
[19]D.Song,J.Li,and Q.Cai,J.Phys.Chem.C 111,18970(2007).
[20]M.Ⅰ.Zaki,B.Vielhaber,and H.Knoezinger,J.Phys. Chem.90,3176(1986).
[21]J.B.Peri,J.Phys.Chem.86,1615(1982).
[22]W.Wu,Z.Wu,C.Liang,X.Chen,P.Ying,and C.Li,J.Phys.Chem.B 107,7088(2003).
[23]A.Y.Khodakov,J.Lynch,D.Bazin,B.Rebours,N. Zanier,B.Moisson,and P.Chaumette,J.Catal.168,16(1997).
[24]A.Muramatsu,T.Tatsumi,and H.Tominaga,J.Phys. Chem.96 1334(1992).
[25]V.Subramani and S.K.Gangwal,Energ.Fuel.22,814 (2008).
[26]K.Xiao,Z.H.Bao,X.Z.Qi,X.X.Wang,L.S.Zhong,K.G.Fang,M.G.Lin,and Y.H.Sun,Chin.J.Catal. 34,116(2013).
[27]M.Zhang,W.Zhang,W.Xie,Z.Qi,G.Wu,M.Lv,S. Sun,and J.Bao,J.Mol.Catal.A 395,269(2014).
[28]X.G.Li,L.J.Feng,L.J.Zhang,D.B.Dadyburjor,and E.L.Kugler,Molecules 8,13(2003).
[29]A.Y.Khodakov,W.Chu,and P.Fongarland,Chem. Rev.107 1692(2007).
[30]N.Kruse,J.Schweicher,A.Bundhoo,A.Frennet,and T.Visart de Bocarm´e,Top Catal.48,145(2008).
[31]M.Xiang,D.Li,W.Li,B.Zhong,and Y.Sun,Catal. Commun.8,503(2007).
[32]T.Ⅰshida,T.Yanagihara,X.H.Liu,H.Ohashi,A. Hamasaki,T.Honma,H.Oji,T.Yokoyama,and M. Tokunaga,Appl.Catal.A 458,145(2013).
[33]G.Jiao,Y.Ding,H.Zhu,X.Li,J.Li,R.Lin,W.Dong,L.Gong,Y.Pei,and Y.Lu,Appl.Catal.A 364 137 (2009).
[34]G.Liu,D.Pan,T.Niu,A.Cao,Y.Yue,and Y.Liu,RSC Adv.5,31637(2015).
[35]Y.P.Pei,J.X.Liu,Y.H.Zhao,Y.J.Ding,T.Liu,W. D.Dong,H.J.Zhu,H.Y.Su,L.Yan,J.L.Li,and W. X.Li,Acs Catal.5,3620(2015).
[36]X.M.Wu,Y.Y.Guo,J.M.Zhou,G.D.Lin,X.Dong,and H.B.Zhang,Appl.Catal.A 340,87(2008).
[37]M.L.Smith,N.Kumar,and J.J.Spivey,J.Phys. Chem.C 116,7931(2012).
Ji-long Zhoua,Wei Xiea,Song Suna∗,Li-li Jia,Li-rong Zhengb,Chen Gaoa,c,Jun Baoa,c∗
a.National Synchrotron Radiation Laboratory,Collaborative Innovation Center of Chemistry for Energy Materials,University of Science and Technology of China,Hefei 230029,China
b.Institute of High Energy Physics,Chinese Academy of Science,Beijing 100039,China
c.CAS Key Laboratory of Materials for Energy Conversion,Department of Materials Science and Engineering,University of Science and Technology of China,Hefei 230026,China
(Dated:Received on March 2,2016;Accepted on April 12,2016)
Activated carbon supported Mo-based catalysts were prepared and reduced under different activation atmospheres,including pure H2,syngas(H2/CO=2/1),and pure CO.The catalysts structures were characterized by X-ray diffraction,X-ray absorption fine structure,and in situ diffuse reflectance infrared Fourier transform spectroscopy.The catalytic performance for the higher alcohol synthesis from syngas was tested.The pure H2treatment showed a high reduction capacity.The presence of a large amount of metallic Co0and low valence state Moφ+(0<φ<2)on the surface suggested a super activity for the CO dissociation and hydrogenation,which promoted hydrocarbons formation and reduced the alcohol selectivity.Ⅰn contrast,the pure CO-reduced catalyst had a low reduction degree.The Mo and Co species at the catalyst mainly existed in the form of Mo4+and Co2+.The syngasreduced catalyst showed the highest activity and selectivity for the higher alcohols synthesis. We suggest that the syngas treatment had an appropriate reduction capacity that is between those of pure H2and pure CO and led to the coexistence of multivalent Co species as well as the enrichment of Moδ+on the catalyst’s surface.The synergistic effects between these active species provided a better cooperativity and equilibrium between the CO dissociation,hydrogenation and CO insertion and thus contributed beneficially to the formation of higher alcohols.
Key words:Higher alcohol synthesis,Activation mechanism,in situ diffuse reflectance infrared Fourier transform spectroscopy,Mo-based catalyst,Syngas
∗
Authors to whom correspondence should be addressed.E-mail: baoj@ustc.edu.cn,suns@ustc.edu.cn
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Electricity Storage With High Roundtrip Efficiency in a Reversible Solid Oxide Cell Stack
- Construction and Evaluation of Merged Pharmacophore Based on Peroxisome Proliferator Receptor-Alpha Agonists
- Solvatochromic Parameters and Preferential Solvation Behavior for Binary Mixtures of 1,3-Dialkylimidazolium Ionic Liquids with Water
- Catalytic Transformation of Oxygenated Organic Compounds into Pure Hydrogen
- Depolymerization of Organosolv Lignin over Silica-alumina Catalysts
- Multiple Plasmonic Resonances and Cascade Effect in Asymmetrical Ag Nanowire Homotrimer