样本数量对白桦群体遗传参数估算的影响
2016-09-23毕志宏魏敏静刘莹莹杨传平魏志刚东北林业大学林木遗传育种国家重点实验室黑龙江哈尔滨150040
毕志宏,魏敏静,刘莹莹,杨传平,魏志刚(东北林业大学 林木遗传育种国家重点实验室,黑龙江 哈尔滨 150040)
样本数量对白桦群体遗传参数估算的影响
毕志宏,魏敏静,刘莹莹,杨传平,魏志刚
(东北林业大学 林木遗传育种国家重点实验室,黑龙江 哈尔滨 150040)
群体遗传参数是群体遗传学研究的主要内容之一,估算值的大小受群体样本数量的影响。以帽儿山试验林场白桦Betula platyphylla种源试验林为对象,采用序列相关扩增多态性(sequence-related amplified polymorphism,SRAP)技术分析了种源内不同样本数量对白桦群体遗传多样性的影响。结果表明:不同样本数量对白桦的估算的各遗传参数有一定影响,但影响各异。其中,对白桦群体多态位点百分率、群体内遗传变异和群体间遗传参数影响显著,对总位点数、多态位点数和基因流影响不显著。样本数量对于群体间与群体内遗传变异的大小的研究结果也产生影响,如样本数量超过8个,显示白桦种源内的遗传变异幅度大于种源间;而样本数量为4个,显示为种源间遗传变异幅度大于种源内。此外,不同样本数量揭示出的种源间亲缘关系不同,如样本数量超过8个,15个种源的聚类结果基本一致;而样本数量为4个,聚类结果无法反映白桦群体的亲缘关系。最后,提出了白桦遗传多样性研究时样本数量的计算公式,认为白桦多态位点百分率达98.5%的样本数量应为11个。图4表3参23
林木遗传育种学;白桦;遗传多样性;样本容量
林木种质资源是陆地生物的避护所和主基因库。林木种质资源的丢失,将引起邻近生物种及其种质以4~13倍的速率丢失[1]。了解林木群体的遗传背景、遗传结构及群体间的亲缘关系,是林木遗传多样研究和采取科学有效措施保护其种质资源的前提和基础[2],也为林木资源的开发与利用提供信息。到目前为止,先后采用不同的标记技术对众多的林木遗传多样性进行了研究[3-6]。群体遗传参数是群体遗传学研究的主要内容之一,估算值的大小受群体样本数量的影响。林木为异交物种,需要一定的数量才能代表1个群体,因此群体内个体数量的多少可能对群体遗传参数估算产生一定影响。然而,长期以来,在树木遗传多样性研究中,群体内个体数量对遗传参数研究结果的影响研究尚无报道。序列相关扩增多态性(sequence-related amplified polymorphism,SRAP)标记建立以来,由于其操作简便、成本低、可信度高、易于测序等特点倍受分子生物学家的青睐[7],被迅速应用到花生Arachis hypogaea,油菜Brassica napus等植物[8-14]的种质资源鉴定评价、遗传图谱构建、重要性状标记以及基因克隆等方面。白桦Betula platyphylla是一种分布范围广、适用性强、用途广泛的阔叶速生树种,然而,有关白桦遗传多样性研究中,同样也没有重视种源内样本数量对遗传参数估算的影响问题[15]。本研究以东北林业大学帽儿山试验林场15个白桦种源为对象,采用SRAP技术分析了白桦种源内不同样本数量对其遗传多样性参数估算的影响,以期确定白桦遗传多样性分析的种源内个体最优数量,也为其他树种遗传多样性研究提供参考。
1 材料与方法
1.1植物材料
试验材料来自东北林业大学帽儿山实验林场内白桦种源试验林,共15个种源,分别为凉水、汪清、新疆、露水河、小北湖、辉南、桓仁、莫尔道嘎、草河口、绰尔、宁夏、清源、东方红、帽儿山、乌伊岭。随机选取个体20个·种源-1,共计300个样本。2012年6月采取当年生嫩叶,用密封袋封好,做好标记,放在冰盒中带回,置于-70℃冰箱中保存,以备提取DNA。
1.2试验方法
1.2.1白桦叶片基因组DNA提取白桦叶片基因组DNA提取参考文献[16]。
1.2.2SRAP标记分析SRAP反应体系为20.00 μL:0.21×106μg·L-1DNA,1.50 μL;25 mmol·L-1Mg2+,1.40 μL;5×16.67 μkat·L-1Taq酶,0.25 μL;2.5 mmol·L-1三磷酸碱基脱氧核苷酸(dNTPs),2.00 μL;10 μmol·L-1引物,0.35 μL。聚合酶链式反应(PCR)反应程序为:94℃预变性5 min,94℃变性1 min,35℃复性1 min,72℃延伸1 min,5个循环;94℃变性1 min,50℃复性1 min,72℃延伸1 min,30个循环,72℃延伸7 min。根据文献[17]中的引物序列,本研究中所用SRAP引物由上海生工合成。从亲缘关系最近的2个种源(莫尔道嘎和桓仁)中分别选5个个体用于多态性引物的筛选。
1.2.3遗传多样性参数的估算15个白桦种源,每个种源按包含4,8,12和20个个体为研究对象,利用筛选出的17对引物对其扩增后分析各项的遗传参数。电泳图谱中的每条带均代表了引物与模板DNA互补的一对结合位点,可记为1个分子标记。根据分子量标准对照反应产物在胶上的对应位置,估计扩增产物的分子量大小,有带的记为 “1”,无带的记为 “0”。所得结果为一二元数据矩阵,利用Popgene 32软件计算各项遗传参数。利用NTsys 2.10e软件分析各源间聚类关系,利用Mintable 15软件分析群体多态位点百分数与群体内个体数量之间的关系和作图。
2 结果与分析
2.1基因组DNA的质量检测及多态性SRAP引物筛选
使用十六烷基三甲基溴化铵(CTAB)法提取的白桦基因组DNA在质量浓度为10.0 g·L-1琼脂糖凝胶电泳成像后如图1所示。样品DNA为1条清晰明亮的条带,无明显拖尾、弥散或其他异常现象。随机抽取部分样品经紫外分光光度计检测,D(260)/D(280)值约1.8。因此可以认为提取的DNA纯度较高,符合SRAP-PCR扩增要求[18]。
前期基于表型研究的白桦种源实验结果表明:莫尔道嘎和桓仁亲缘关系最近,因此,本研究中,从这2个种源内各随机选择5个个体用于引物的筛选。以条带清晰并且群体与个体间有差异条带的引物为选择标准,从合成的30对引物中进行筛选。图2中5个泳道为1组扩增结果,从左开始分别为莫尔道嘎和桓仁的种源及个体,图2为引物me2em3组合扩增结果。从图2可看出:该引物扩增时,不仅2个种源间存在差异性条带,且同一种源内不同个体间也有清晰差异条带。这表明引物me2em3符合引物选择的标准,可用于白桦群体遗传多样性的研究。按此标准,从合成的30对引物中,筛选出17个条带清晰、多态性高的引物用于实验分析(表1)。
图1 部分白桦样本基因组DNA的琼脂糖凝胶电泳检测结果Figure 1 Detection of Betula platyphlla genomic DNA using agarose gel electrophoresis
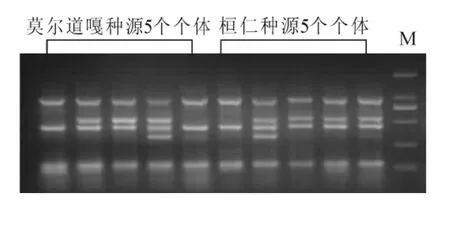
图2 白桦SRAP引物(me2em3)的筛选Figure 2 Primer selected for SRPA of B.platyphlla

表1 SRAP引物序列Table 1 primer sequences of SRAP
2.2白桦种源不同样本数量估算的群体遗传多样性
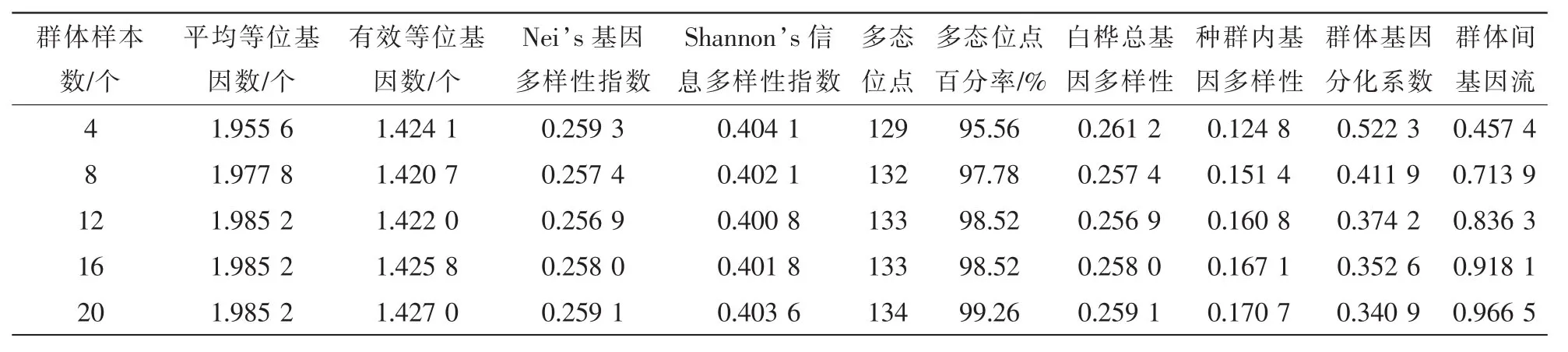
表2 不同样本数量估算的遗传参数比较Table 2 Comparison of different sample size estimation for genetic parameters
利用17对SRAP引物分别对包含4,8,12和20个样本个体的白桦共15个种源进行扩增后分析,并利用POP gene软件计算各遗传参数。表2为不同样本数量时白桦群体遗传多样性各参数的平均值。结果发现:样本数量不同时,各项遗传参数平均数也存在一定差异。这说明样本数量对白桦群体的遗传参数估算有一定的影响。如当群体样本数量分别为4,8,12,16和20时,各群体的多态位点分别为129,132,133,133和134个,多态位点百分率为95.56%~99.26%。平均等位基因数为1.955 6~1.985 2,有效等位基因数为1.420 7~1.427 0,Nei's指数的变动范围为0.256 9~0.259 3,Shannon指数的变动范围为0.400 8~0.404 1。进一步分析发现,在不同样本数量条件下,各项遗传参数变异最大种源均为凉水,如当群体样本数量为 4时,共检测到 135个位点,其中多态位点有 129个,多态位点百分率达95.56%。多态位点百分率为 17.78%~52.33%,平均等位基因数为1.177 8~1.533 3,有效等位基因数为1.112 6~1.356 6,Nei's指数的变动范围为0.066 0~0.205 9,Shannon指数的变动范围为0.098 3~0.304 2,上述的遗传参数均表现为凉水种源各项遗传参数最大,且在15个种源间变化趋势一致。当群体样本数为8,12,16,20时凉水种源各项遗传参数均最大。而各项遗传参数最小的种源随样本数量的变异均不同,如样本数量为12时,共检测到135个位点,其中多态位点有133个,多态位点百分率达98.52%,多态位点百分率为34.07%~77.78%,平均等位基因数为1.340 7~1.777 8,有效等位基因数为1.166 7~1.420 9,Nei's指数为0.101 5~0.249 2,Shannon指数为0.156 6~0.378 5。上述遗传参数中,汪清种源各项遗传参数最小;而样本数量为16时,共检测到135个位点,其中多态位点有 133个,多态位点百分率达98.52%,种源的多态位点数为47~108,多态位点百分率为34.81%~80.00%,平均等位基因数为1.348 1~1.800 0,有效等位基因数为1.180 0~1.425 4,Nei's指数为0.108 1~0.251 1,Shannon指数为0.165 0~0.381 2,上述遗传参数中,清源种源的各项遗传参数最小;样本数量为8和4时,各项遗传参数最小的为淖尔种源。此外,4个样本数量与其他样本数量之间的平均等位基因数,多态位点数,多态位点百分率,Ht,Hs,Gst和Nm差异较大。
表2同时也表明:样本数量对白桦群体内与群体间遗传变异大小的研究结果也有影响。如当群体样本数量为4时,47.78%的遗传变异存在于种源内,52.22%的遗传变异存在于种源间。当群体内样本数量超过8时,研究反映出白桦种群内遗传变异幅度超过群体间;如群体样本数量为8时,58.82%的遗传变异存在于种源内,41.18%的遗传变异存在于种源间;当群体样本数量为20时,65.88%的遗传变异存在于种源内,34.12%存在于种源间。
2.3不同样本数量对群体间亲缘关系分析结果的影响
为了进一步分析种源内样本数量差异对于白桦遗传结构研究结果的影响,笔者又研究了不同样本数量下的白桦种源遗传进化关系。图3表明:样本数量对于白桦种源遗传进化关系分析结果有一定的影响。如种源样本数量超过12时,15个种源的聚类结果基本一致(图3 C,图3D和图3E),均可在遗传距离为0.17时,分成4个类群,而且每个类群包括的种源基本一致。样本数量为8时,在0.17的遗传距离处,15个种源则划分为5个类群,其中凉水种源单独聚成一类(图3B)。而种源样本数量为4时,在0.17的遗传距离处,15个种源被划分成7个类群,反映不出群体进化的基本规律(图3A)。此时,只有当遗传距离达到0.25左右时,15个种源才可划分成4类,但凉水种源单独聚成一类,帽儿山、东方红和乌伊岭种源聚成一类,新疆和露水河种源聚成一类,其余9个种源聚成一类。由此可见:当样本数量仅有4个时,其揭示的白桦群体间的亲缘关系与样本数量为8个和超过12时存在差异。而且,种源样本数量为4时,15个种源遗传距离变化范围为0.050 6~0.280 0,其中清源和宁夏种源的遗传距离最小,桓仁和乌伊岭种源的遗传距离最大。当种源样本数量为分别8,12,16和20,15个种源间在0.044 0~0.212 5范围内变化,而此时淖尔和凉水种源间的遗传距离最大,莫尔道嘎和桓仁种源的遗传距离最小。
2.4样本数量和主要遗传参数的相关分析
由于样本数量对白桦遗传参数与遗传结构的分析结果均产生一定的影响,因此为弄清样本数量与各遗传参数之间的具体关系,分析了不同样本数量与其对应的各项遗传参数作相关性。结果(表3)表明:种源内不同样本数量与白桦群体多态位点百分率、群体内遗传变异和群体间遗传变异参数相关性显著,与总位点数、多态位点数和基因流的相关性不显著。这进一步表明,在研究异交群体遗传多样性时,应科学估算出群体样本的最佳数量。
2.5白桦遗传多样性研究时样本数量的确定

表3 样本容量与主要遗传参数的相关性Table 3 Correlation between sample size and main genetic parameters
白桦种源样本数量与一些遗传参数之间存在显著或极显著相关性,这为确定白桦遗传多样性研究时,确定合理的样本数量提供了可能。在群体遗传多样性研究中,多态位点百分率越高越有利于分析和研究群体遗传多样和遗传结构,因此对白桦种源不同样本数量与多态位点百分率进行拟合分析(图4),在95%的置信区间内,两者之间呈一元二次曲线关系,决定系数为89%,表明两者之间存在较好的相关性。我们在白桦遗传多样性研究时,在综合考虑成本、技术难度等基础上,根据所需多态位点百分率大小,计算出所需样本的数量。如当白桦遗传多样性研究时,以最低所需多态位点百分率98.5%为标准,则每个种源所需样本数量为10.673个,实际操作中可设为11个样本个体。
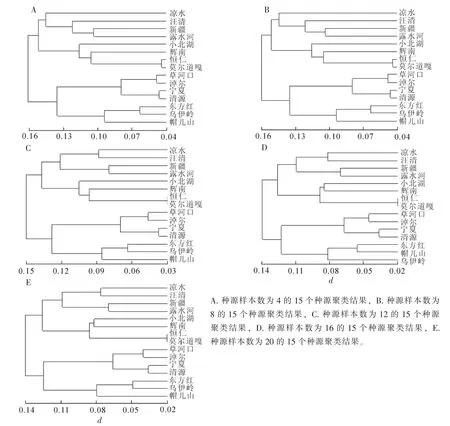
图3 不同样本数量时15个白桦种源聚类图Figure 3 Different number of samples for 15 birch provenance clustering map
3 讨论与结论
对于异交植物,种群内和种群间遗传变异较大,必须有一定数量个体才能代表该种群的基因库,因此群体样本数量会对研究结果产生影响。如HENSEN等[19]在研究白鲜Dictamnus albus时发现,其遗传多样性及多态位点随群体样本容量的不同而发生变化,而且群体大小、多态位点百分率和遗传多样性间存在显著的相关性。本研究表明:白桦种源内样本数量对白桦群体多态位点百分率、群体内遗传变异和群体间遗传参数影响显著,对总位点数、多态位点数和基因流影响不显著。ZHAO等[20]研究发现不同样本容量的野生大豆Glycine soja种群间得到的遗传多样性指数存在较大差异。本研究同样发现:白桦种源样本数量不同时,其估算的遗传多样性指数也存在一定差异。如样本数量为4时,多态位点数和百分率最大与最小的种源分别为凉水和淖尔,而样本数量超过8个时,则为凉水和清源。此外,种源内不同个体数量反映的种源内与种源间的遗传变异不同,如种源内样本数为4时,白桦种源间的遗传变异大于种源内。而当群体样本数量超过8时,白桦种源内的遗传变异幅度大于种源间。
本研究发现:群体样本数量对遗传群体间遗传距离的分析结果产生一定的影响,如源样本数量为4时,清源和宁夏种源的遗传距离最小,桓仁和乌伊岭种源的遗传距离最大。而样本数量超过8时,淖尔和凉水的遗传距离最大,莫尔道嘎和桓仁的遗传距离最小。这与样本数量超过10时,通过表型分析得到的研究结果相同,如高玉池等[21]利用表型性状对帽儿山地区10年生白桦种源间的亲缘关系进行了分析,结果表明淖尔和凉水的遗传距离最大,莫尔道嘎和桓仁的遗传距离相对较小。此外,样本数量也对白桦种源间的遗传进化关系分析结果有一定的影响,如在相同的遗传距离处(0.17),样本数量为4时,15个种源被划分成7个类群;样本数量为8时,15个种源被划分成5个类群;样本数量超过12时,15个种源被划分成4个类群。上述分析表明:群体样本数量不仅对白桦各群体的遗传参数产生影响,也对群体间遗传进化关系的分析结果有一定的影响。

图4 白桦种源样本数量与多态位点百分率的关系Figure 4 Relationship between Betula platyphylla provenances samples and the percentage of polymorphic loci
异交植物多少个体才能代表一个群体,才能得到较为客观真实的遗传多样性参数在草本植物或花卉植物中有所研究。如SINGH等[22]在研究野生二粒小麦Triticum dicoccoides的遗传多样性时发现,有的群体5~6个个体就可以使多样性标准差不大于10%,个别一些则需12个以上的个体。JULIO等[23]发现,蔷薇Rosaceae种群个体数超过10个以后,就不能提供更多的种群杂合度信息。本研究发现:白桦种源内样本的数量与其多态位点百分率呈一元二次曲线相关,且决定系数达89%。并以此为公式计算白桦遗传多样性研究时,如设定要探测到的多态位点百分率达98.5%以上时,每个种源至少必须含有11个样本个体。
[1]魏志刚,高玉池,刘桂丰,等.白桦核心种质初步构建[J].林业科学,2009,45(10):74-80. WEI Zhigang,GAO Yuchi,LIU Guifeng,et al.Preliminary construction of core collection of Betula platyphylla germplasm[J].Sci Silv Sin,2009,45(10):74-80.
[2]顾万春.中国林木种质资源保存研究与对策[C]//陈宜瑜.中国生物多样性保护与研究进展:第5届全国生物多样性保护与持续利用研讨会论文集.北京:气象出版社,2004:93-105.
[3]罗建勋,王国良,辜云杰,等.杉木优树半同胞子代测定及优良家系选择初步研究[C]//中国林学会.第2届中国林业学术大会:S2功能基因组时代的林木遗传与改良论文集.南宁:[s.n.].2009:671-673.
[4]李斌,顾万春.松属植物遗传多样性研究进展[J].遗传,2003,25(6):740-748. LI Bin,GU Wanchun.Review on genetic diversity in Pinus[J].Hereditas,2003,25(6):740-748.
[5]张萍,周志春,金国庆,等.木荷种源遗传多样性和种源区初步划分[J].林业科学,2006,42(2):38-42. ZHANG Ping,ZHOU Zhichun,JIN Guoqing,et al.Genetic diversity analysis and provenance zone allocation of Schima superba in China using RAPD markers[J].Sci Silv Sin,2006,42(2):38-42.
[6]庄振宏.运用RAPD技术分析长叶榧(Torreya jackii Chun)的遗传多样性[D].福州:福建师范大学,2002. ZHUANG Zhenhong.Genetic Diversity Analysis by RAPD on Torreya jackii Chun[D].Fuzhou:Fujian Normal University,2002.
[7]谭碧玥,王源秀,徐立安.分子标记SRAP及其在林木研究中的应用[J].世界林业研究,2009,22(5):45-50. TAN Biyue,WANG Yuanxiu,XU Li'an.Molecular marker SRAP and its application in forest research[J].World For Res,2009,22(5):45-50.
[8]刘月光,滕永勇,潘辰,等.应用SRAP标记对莲藕资源的聚类分析[J].氨基酸和生物资源,2006,28(1):29-32. LIU Yueguang,TENG Yongyong,PAN Chen,et al.Cluster analysis of Nelumbo based on SRAP markers[J].AminoAcid Biol Resour,2006,28(1):29-32.
[9]史红丽,韩明玉,赵彩平.桃遗传多样性的SRAP和SSR标记分析[J].华北农学报,2009,24(6):187-192. SHI Hongli,HAN Mingyu,ZHAO Caiping.Genetic diversity of Prunus persica using SRAP and SSR markers[J]. Chin J North China,2009,24(6):187-192.
[10]陈静,胡晓辉,苗华荣,等.花生SRAP-PCR技术体系的优化[J].山东农业科学,2008,10(5):24-26. CHEN Jing,HU Xiaohui,MIAO Huarong,et al.Optimization of SRAP-PCR reaction system on peanut[J].Shandong Agric Sci,2008,10(5):24-26.
[11]张羽,李英,许伟,等.利用SRAP和SSR标记对汉中地区主要油菜品种的遗传多样性分析[J].四川农业大学学报,2013,31(4):370-376. ZHANG Yu,LI Ying,XU Wei,et al.Analysis of genetic diversity of Brassica napus mainly popularized in Hanzhong based on SRAP and SSR markers[J].J Sichuan Agric Univ,2013,31(4):370-376.
[12]王惠哲,李淑菊,管炜.应用SRAP标记分析黄瓜的遗传差异[J].生物技术通报,2009(2):76-79. WANG Huizhe,LI Shuju,GUAN Wei.Analysis on the genetic relationship of four types of cucumber germphasms by SRAP markers[J].Biotechnol Bull,2009(2):76-79.
[13]张凯,罗小敏,蒋玉春,等.甘薯种质资源的SRAP鉴定及遗传多样性分析[J].核农学报,2013,27(5):568-575. ZHANG Kai,LUO Xiaomin,JIANG Yuchun,et al.Genetic diversity among main sweeppotato resources using SRAP markers[J].J Nucl Agric Sci,2013,27(5):568-575.
[14]李朋飞,霍秀爱,程永强,等.基于SRAP的西瓜种质资源遗传多样性评价[J].中国农业科技导报,2013,15(2):89-96. LI Pengfei,HUO Xiuai,CHENG Yongqiang,et al.Assement of genetic diversity in watermelon based on SRAP analysis[J].J Agric Sci Technol,2013,15(2):89-96.
[15]姜静,杨传平,刘桂丰,等.应用RAPD技术对东北地区白桦种源遗传变异的分析[J].东北林业大学学报,2001,29(2):30-34. JIANG Jing,YANG Chuanping,LIU Guifeng,et al.Analysis on genetic variation within and among Betula platyphylla provenances in northeast China using RAPD markers[J].J Northeast For Univ,2001,29(2):30-34.
[16]詹亚光,曾凡锁.富含多糖的白桦成熟叶片DNA的提取方法[J].东北林业大学学报,2005,33(3):24-25. ZHAN Yaguang,ZENG Fansuo.A method of DNA extraction from mature birch leaves rich in polysaccharide[J].J Northeast For Univ,2013,33(3):24-25.
[17]任娜.红豆杉内生真菌遗传多样性分析及生物转化研究[D].长沙:中南大学,2011. REN Na.Genetic Diversity Analysis and Biotransformation Research among Endophytic Fungi from Taxus[D]. Changsha:Central South University,2011.
[18]张景阁.金磁微粒纯化细菌基因组DNA方法的研究[D].西安:西北大学,2011. ZHANG Jingge.Study on the Genomic DNA Method by Goldmag Particles[D].Xi'an:Northwestern University,2011.
[19]HENSEN I,OBERPRIELER C.Effects of population size on genetic diversity and seed production in the rare Dictamnus albus(Rutaceae)in central Germany[J].Conserv Genet,2005,6(1):63-73.
[20]ZHAO Ru,CHENG Zhou,LU Weifeng,et al.Estimating genetic diversity and sampling strategy for a wild soybean (Glycine soja)population based on different molecular markers[J].Chin Sci Bull,2006,51(10):1219-1227.
[21]高玉池,魏志刚,杨传平,等.帽儿山地区10年生白桦种源试验[J].浙江林学院学报,2009,26(6):784-791. GAO Yuchi,WEI Zhigang,YANG Chuanping,et al.A 10-year-old provenance trial of Betula platyphylla in the Maoershan area of Heilongjiang Province[J].J Zhejiang For Coll,2009,26(6):784-791.
[22]SINGH M,CHABANE K,VALKOUN J,et al.Optimum sample size for estimating gene diversity in wild wheat using AFLP markers[J].Genet Resour Crop Evol,2006,53(1):23-33.
[23]JULIO N,SOBRAL A,BUEÑAS J R,et al.RAPD and ISSR markers indicate diminished gene flow due to recent fragmentation of Polylepis australis woodlands in central Argentina[J].Biochem Syst Ecol,2008,36(5/6):329-335.
Genetic parameters of Betula platyphylla provenances due to sample size
BI Zhihong,WEI Minjing,LIU Yingying,YANG Chuanping,WEI Zhigang
(State Key Laboratory of Forest Genetics and Breeding,Northeast Forestry University,Harbin 150040,Heilongjiang,China)
Population genetic parameters,one of the main constituents of population genetics,and the size of estimated values are influenced by the number of population samples.In this study Betula platyphylla (birch)from Cap Mountain Experimental Forest Farm's Provenance Testing Forest was chosen as the experimental object and sequence related amplified polymorphism(SRAP)technology was used for analysis.Results showed that genetic parameters with different sample sizes had different effects on birch.Genetic variation,population of birch groups,and the percentage of polymorphic numbers within populations influenced genetic parameters.Influence from genetic variation and genetic parameters between groups was strong,but total numbers,polymorphic numbers,and gene flow were not affected.When the sample size was greater than eight,genetic variation amplitude of the birch source was greater than that of the provenances;when the sample size was four,genetic variation was greater than the source of genetic variation among provenances for different samples.For genetic relationships of different provenances with sample size greater than eight,15 kinds of source clustering results were basically the same.However,with a sample size of four,clustering results did not reflect the evolutionary relationship between birch groups.Also,11 birch samples were found to have a polymorphic number that was 98.5%.[Ch,4 fig.3 tab.23 ref.]
forest tree breeding;Betula platyphylla;genetic diversity;sample capacity
S722.3
A
2095-0756(2016)04-0564-07
10.11833/j.issn.2095-0756.2016.04.003
2015-06-10;
2015-10-09
国家高技术研究发展计划(“863”计划)项目(2011AA100202)
毕志宏,从事木材次生生长等研究。E-mail:bizhihong9191@163.com。通信作者:魏志刚,教授,博士,从事林木遗传育种等研究。E-mail:zhigangwei1973@163.com
