茶树咖啡碱合成酶CRISPR/Cas9基因组编辑载体的构建
2016-09-21唐雨薇刘丽萍王若娴陈宇宏刘仲华刘硕谦
唐雨薇,刘丽萍,王若娴,陈宇宏,刘仲华,2,3,刘硕谦,2,3*
1.湖南农业大学园艺园林学院,湖南 长沙 410128;2.国家植物功能成分利用工程技术研究中心,湖南 长沙 410128;3.教育部茶学重点实验室,湖南 长沙 410128
茶树咖啡碱合成酶CRISPR/Cas9基因组编辑载体的构建
唐雨薇1,3,刘丽萍1,3,王若娴1,陈宇宏1,刘仲华1,2,3,刘硕谦1,2,3*
1.湖南农业大学园艺园林学院,湖南 长沙 410128;2.国家植物功能成分利用工程技术研究中心,湖南 长沙 410128;3.教育部茶学重点实验室,湖南 长沙 410128
CRISPR/Cas9技术是一门新兴的基因组定点编辑技术,具有操作简单、高效的优点,可轻松实现对目标基因的敲除、替换和定点突变等操作。该技术刚诞生,就受到了全球生命科学领域研究者的关注,不到 3年的时间就已经成功应用于多种动、植物当中。然而 CRISPR/Cas9技术在茶树中的应用面临载体构建问题,本文以茶树咖啡碱合成酶为例,联合采用常规PCR、Overlapping PCR和Golden Gate Cloning技术,构建了包含茶树咖啡碱合成酶双靶点的CRISPR/Cas9基因编辑载体,为CRISPR/Cas9介导的基因组编辑技术在茶树中的应用奠定了坚实基础。
茶树;基因组编辑技术;咖啡碱合成酶;CRISPR/Cas9技术
虽然我国一直非常重视茶树的品种改良工作,但由于茶树具有生长周期长、自交不亲和的特性,使得采用常规育种技术难以实现茶树育种工作取得突破性进展。近年来分子育种及基因工程等现代分子生物学技术在水稻、西红柿、马铃薯等农作物品种改良中发挥了重要作用,也为茶树育种提供了新的途径。因而,茶树分子生物学研究成了茶叶科学中最活跃和进展最快的领域。最近,茶树转录组测序工作取得了重要进展[1-4]。目前,GenBank已收录了约35万条茶树mRNA序列(http://www.ncbi.nlm.nih.gov/nuccore/?term=Camellia+ sinensis),几乎涵盖了茶树中所有功能基因编码序列。此外,我国茶树的全基因组测序工作也于几年前在多家单位开始启动,并已相继完成,茶树全基因组序列将会在短期内释放[5]。然而,面对大量的基因序列,我们对其功能了解却是非常缺少,制约了基因资源的利用。因此,确定茶树新基因的功能并高效利用新基因将是未来茶树分子生物学研究中一个非常重要的主题,也就是说茶树后基因组时代即将来临。随着现代分子生物技术的突飞猛进,新型基因组编辑技术CRISPR/Cas9(Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9)将会在茶树功能基因研究中发挥重要作用,预计将成为茶树后基因组时代不可或缺的研究工具。
基因组定点编辑技术是分子育种、基因的功能研究和遗传改造等重要工具,是通过某种途径对基因组DNA特定位点进行改造的一种手段。迄今为止,应用时间比较长的基因组编辑技术有锌指核酸酶(Zinc-finger nucleases,ZFN)[6]和类转录激活因子效应物核酸(Transcription activator-likeeffector nucleases,TALEN)技术[7]。但由于这两项技术DNA结合结构域的改造较为复杂,对每一个基因位点的编辑都需要重新设计、合成和组装2个核酸酶,载体构建困难,成为限制其发展的瓶颈[8]。科学家一直在寻找精确而简单的基因组编辑方法,直到 2013年发现了CRISPR/Cas9技术[9]。CRISPR/Cas9技术是通过一段RNA来识别靶位点,因而在设计和构建上更加简单,只需合成一个sgRNA就能实现对基因的特异性编辑[10]。自 CRISPR/Cas9技术的诞生到现在,短短几年内该技术迅速应用于世界各地的实验室,并成为基因组定点编辑的首选方法[11]。
CRISPR/Cas广泛存在于古生菌和细菌的基因组中,属自身免疫系统,可降解入侵的病毒或质粒 DNA[12]。在该免疫系统中,Cas蛋白(CRISP-associated protein)含有两个核酸酶结构域,可以分别切割两条DNA链,切割后DNA双链断裂从而使入侵的外源DNA降解[13]。CRISPR/Cas系统的组成结构比较固定,由 CRISPR序列与 Cas基因家族组成,其中CRISPR由一系列高度保守的重复序列(Repeat)与间隔序列(Spacer)相间排列组成,间隔序列可特异识别外源 DNA[13]。在CRISPR序列附近存在高度保守的CRISPR相关基因,这些基因编码的蛋白具有核酸酶功能,可以对DNA序列进行特异性切割[14]。
CRISPR系统分为3种类型,现在常用的CRISPR/Cas系统由类型Ⅱ系统改造而来。类型Ⅱ系统的特征性蛋白为Cas9蛋白,具有加工产生 CRISPR RNA(crRNA)和切割双链DNA功能。crRNA(CRISPR-derived RNA)通过碱基配对与 tracrRNA(trans-activating RNA)结合形成tracrRNA/crRNA复合体,此复合体一旦形成就能引导核酸酶Cas9蛋白在与crRNA配对的序列靶位点剪切双链DNA。而tracrRNA/crRNA复合体可通过人工设计,融合crRNA与tracrRNA形成sgRNA(single-guided RNA),sgRNA足以引导Cas9 对DNA的定点切割(图1)[13,15]。sgRNA可以通过载体表达或者化学合成后与Cas9蛋白共同进入细胞,对特异DNA序列剪切,从而促使DNA 发生NHEJ(nonhomologousend-joining)导致的基因缺失或同源重组,实现基因敲除[16]。CRISPR-Cas9 体系的RNA-DNA识别机制为选择性基因组编辑提供了一个简便而强大的工具。来自Streptococcus pyogenes的Cas9由于识别序列仅为2个碱基(GG),几乎可以在所有的基因中找到大量靶点,因此得到广泛的应用。Cas9蛋白在目前测试过的几乎所有生物和细胞中均有活性,包括细菌、酵母、植物、鱼,以及哺乳动物细胞[17]。该体系其中一个最重要的优势是Cas9蛋白可在多个不同的sgRNA的引导下同时修饰多个基因组靶点。

图1 CRISPR/Cas9工作原理[15]Fig.1 The working model of the CRISPR/Cas9 system[15]
目前植物CRISPR/Cas9系统已日趋完善,已在十多种植物中成功实现了定点基因组编辑,除了在本氏烟草[18-19]、拟南芥[20]等模式植物上获得了成功,很快在小麦[21]、玉米[22]、水稻[23]、高粱[19]、西红柿[24]以及甜橙[25]等农作物上也成功实现了 CRISPR/Cas9的应用。最近,先后利用 CRISPR/Cas9技术实现了木本植物杨树[26]和观赏植物牵牛花[27]基因组的定点编辑,将 CRISPR/Cas9系统在植物中应用推上了新的领域。
然而,到目前为止尚未见有关CRISPR/Cas9技术在茶树上的应用报道,其中一个重要的原因是针对茶树的 CRISPR/Cas9基因编辑载体构建技术尚不完善。本文以茶树中咖啡碱合成酶为例,建立茶树CRISPR/Cas9基因编辑载体的构建方法,为 CRISPR/Cas9基因编辑技术在茶树中的应用奠定理论基础。
1 材料与方法
1.1质粒、引物、菌种与试剂
植物 CRISPR/Cas9基因编辑载体为pYLCRISPR/Cas9P35S-H,GenBank登录号为AY310901,是在双元载体质粒pCAMBIA1300(AF234296)的基础上进行改造而来的,由花椰菜花叶病毒(CaMV)35 S启动子驱动Cas9p序列的表达。该质粒在E.coli TOP10F′(LacIq)菌株繁殖,该菌株LacIq基因型产生的阻碍蛋白可抑制ccdB大肠杆菌致死基因的表达。植物CRISPR/sgRNA载体[28]pYLgRNAAtU3d-LacZ和 pYLgRNA-AtU3b,为植物CRISPR/Cas9基因编辑载体的中间载体,分别提供small nuclear(sn)RNA U3d和U3b启动子序列,驱动sgRNA的表达并保证转录出的sgRNA停留在细胞核中与Cas9结合,且二者均提供长度为57 nt的sgRNA3′区域保守结构序列,可与靶基因的 20 nt靶序列组装成sgRNA。此外,pYLgRNA-AtU3d-LacZ还提供了 LacZ标记基因(198 bp)的表达元件,可在含有X-gal的培养基产生蓝色菌斑筛选阳性克隆。以上质粒均由华南农业大学刘耀光教授实验室惠赠。
本文所需引物序列如表1所示,引物合成与序列测序均由上海生工生物工程技术服务公司完成。发根农杆菌 GV3101和大肠杆菌DH5α为本实验室保存。PCR纯化试剂盒与质粒提取试剂盒菌购自全式基因生物有限公司,KOD-Plus酶购自东洋纺(上海)生物科技有限公司,BsaI-HF酶和 T4 DNA连接酶购自New England Biolabs有限公司,其余分子生物学试剂和试剂盒均购自上海生工生物工程技术服务公司。

表1 引物序列Table 1 The list of primer sequences
1.2sgRNA序列设计及分析
为提高打靶效率针对TCS基因设计2个靶点,设计靶点遵循如下原则:目的基因的正链和负链靶点的打靶效率大致相同,都可以设计靶点;靶点的GC%尽量不要低于40%,靶点序列GC%偏高(50%~70%)有较高的打靶效率;靶点内(按5-N20NGG-3方向)不要有连续4个以上的T,以防RNA Pol III将其作为转录终止信号;虽然非特异打靶(脱靶)对植物基因打靶不是很重要的问题,但应进行靶点特异性分析,用靶点+NGG(前后各加几十碱基)与茶树转录组做 blast分析(参数选择Somewhat similar sequences),避免使用与同源序列差异少于5个碱基的靶点(在切点附近和PAM有2个碱基差异就具有特异性)。把靶点序列连接到 sgRNA序列的 5′端(target sequence + GTTTTAGAGCTAGAAATAGCA AGTTAAAATAAGGCTAGTCCGTTATCAAC TTGAAAAAGTGGCACCGAGTCGGTGCTTT TTTT),利用在线软件 RNA Folding Form(http://mfold.rna.albany.edu/?q=mfold/RNA-F olding-Form2.3)做二级结构分析。
1.3sgRNA表达盒构建方法
1.3.1Target 1 sgRNA(T1sgRNA)构建
首先进行第一轮PCR,第一轮PCR进行2个反应。取2~5 ng pYLgRNA-AtU3d-LacZ质粒为模板,在1个PCR反应中使用引物 U-F 和TCSU3dT1;第2个PCR反应中使用引物TCSgRT1和gR-R。第1个PCR和第2个PCR反应分别在两个 PCR管中进行,采用相同的PCR反应体系(表2)和相同的PCR反应程序:94℃预变性 2 min,25~28循环,94℃ 10 s,58℃ 15 s,68℃ 20 s。取 3 μL反应液进行电泳,检查产物长度是否符合预期,并估计样品的大致浓度。第一轮PCR反应完成后,进行第二轮PCR。取第一轮PCR产物U3dT1-gRNA 各1 μL用H2O稀释10倍,取1 μL为模板,以 Pps-R-C和 Pgs-GG2为引物,扩增相应的U3dT1-gRNA,采用表3所示的反应体系。PCR反应程序:94℃预变性2 min,17~20个循环,94℃ 10 s,58℃ 15s,68℃ 20 s。取 3 μL反应液进行电泳,检查产物长度是否符合预期,并估计样品的大致浓度。
1.3.2Target 2 sgRNA(T2sgRNA)构建
首先进行第一轮 PCR,同样进行 2个反应。取2~5 ng pYLgRNA-AtU3b质粒为模板,在第 1个 PCR反应中使用引物 U-F和TCSU3bT2,第 2个 PCR反应使用引物TCSgRT2和gR-R。U-F/TCSU3bT2扩出U3bT2序列,gR-R/TCSgRT2扩出T2sgRNA序列。采用与构建T1sgRNA第一轮PCR反应相同的PCR反应体系和PCR反应程序。取3 μL反应液进行电泳,检查产物长度是否符合预期,并估计样品的大致浓度。第一轮PCR完毕,进行第二轮PCR。取第一轮PCR产物U3bT2-gRNA各1 μL用H2O稀释10倍,取1 μL为模板,以Pps-GG2和Pgs-L-C为引物,扩增相应的 U3bT2-gRNA。采用与构建T1sgRNA第二轮PCR反应相同的PCR反应体系和PCR反应程序。取3 μL反应液进行电泳,检查产物长度是否符合预期。
1.4组装sgRNA表达盒到pYLCRISPR/Cas9载体
1.4.1双元载体与 sgRNA表达盒的酶切-连接反应
采用PCR产物纯化试剂盒对T1sgRNA和T2sgRNA PCR产物纯化,使用表4所示的酶切-连接反应体系,反应程序:37℃,1 min,16℃,1 min,30个循环;最后55℃灭活内切酶 5 min。以PCR产物纯化试剂盒纯化酶切-连接产物。

表2 第一轮PCR反应体系Table 2 The PCR reaction system for the first round of PCR

表3 第二轮PCR反应体系Table 3 The PCR reaction system for the second round of PCR
1.5酶切-连接产物的转化与质粒提取
从-80℃冰箱中取出50 μL DH 5α感受态细胞,放置在冰上直至融化,取10 μL连接产物加入DH 5α感受态细胞离心管,轻轻小弹离心管,使之混合均匀,冰浴20 min,在精确的 42℃水浴中热击 90 s,迅速将管子移到冰中放置2 min,加入平衡至室温的750 μL LB培养基,37℃振荡培养45 min,取100 μL菌液涂到含50 μg·mL-1Kan、0.5 mmol·L-1IPTG 和40 μg·mL-1X-gal的LB平板,将平板于37℃倒置过夜培养,挑取蓝斑菌落,于3 mL含50 μg·mL-1Kan的LB液体培养基中震荡培养过夜,使用质粒提取试剂盒提取质粒,使用Mlu I酶切验证,挑取正确质粒进行测序验证。
2 结果与分析
2.1sgRNA设计
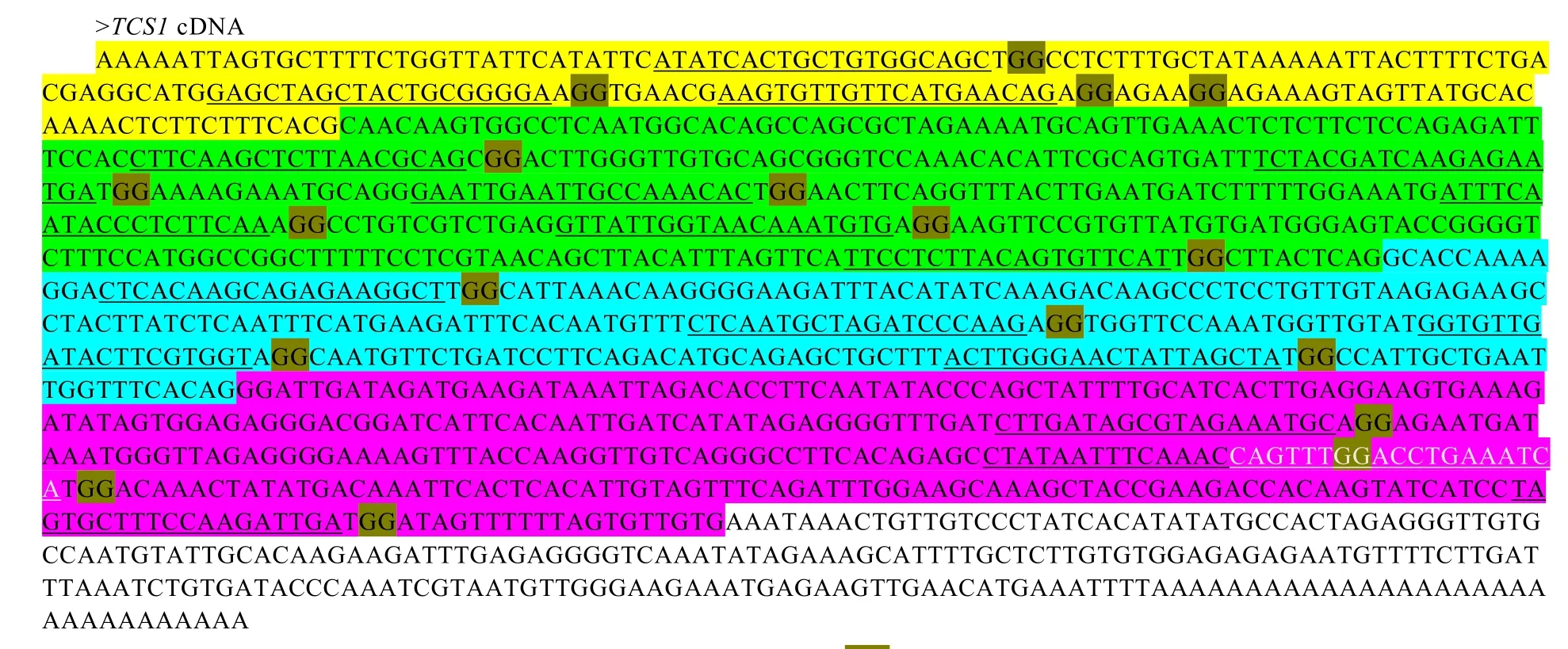
sgRNA序列全长 97 nt,分为两部分,5′决定靶序列的19 nt种子序列(Seed sequence)和3′区域为保守的结构序列。因此构建针对特定靶位点的 sgRNA,只需要克隆决定靶序列的5′端20 nt。从GenBank中下载茶树TCS基因组序列和cDNA序列,分析其内含子所在区域。结果表明(图 2),TCS1基因含 3个内含子。在此基础上搜寻 TCS的 CRISPR/Cas9靶序列,结果共搜寻到18个不含内含子的候选靶序列。利用RNA Folding Form对18个候选靶序列进行结构分析,靶序列与sgRNA序列产生连续配对 7 bp以上会抑制其与染色体DNA靶序列结合靶点,因此要避免使用连续配对7 bp以上的靶序列。分析结果表明,在TCS1基因 ORF内的靶序列“CTCACAAGCAGA GAAGGCT”(设为靶序列T1)和非编码区内靶序列“ATATCACTGCTGTGGCAGC”(设为靶序列T2)较为理想,可作为sgRNA序列的种子序列。

表4 双元载体与sgRNA表达盒的酶切-连接反应体系Table 4 The reaction system for the digestion and ligation of sgRNA expression cassettes and pYLCRISPR/Cas9 vector
2.2质粒提取
将分别含有 pYLgRNA-AtU3d-LacZ、pYLgRNA-AtU3b和pYLCRISPR/Cas9P35S-H质粒的菌划线活化,挑取单菌摇菌,采用质粒提取试剂盒(小提)提取质粒。电泳结果表明(图3),提取的质粒符合下一步实验的要求。
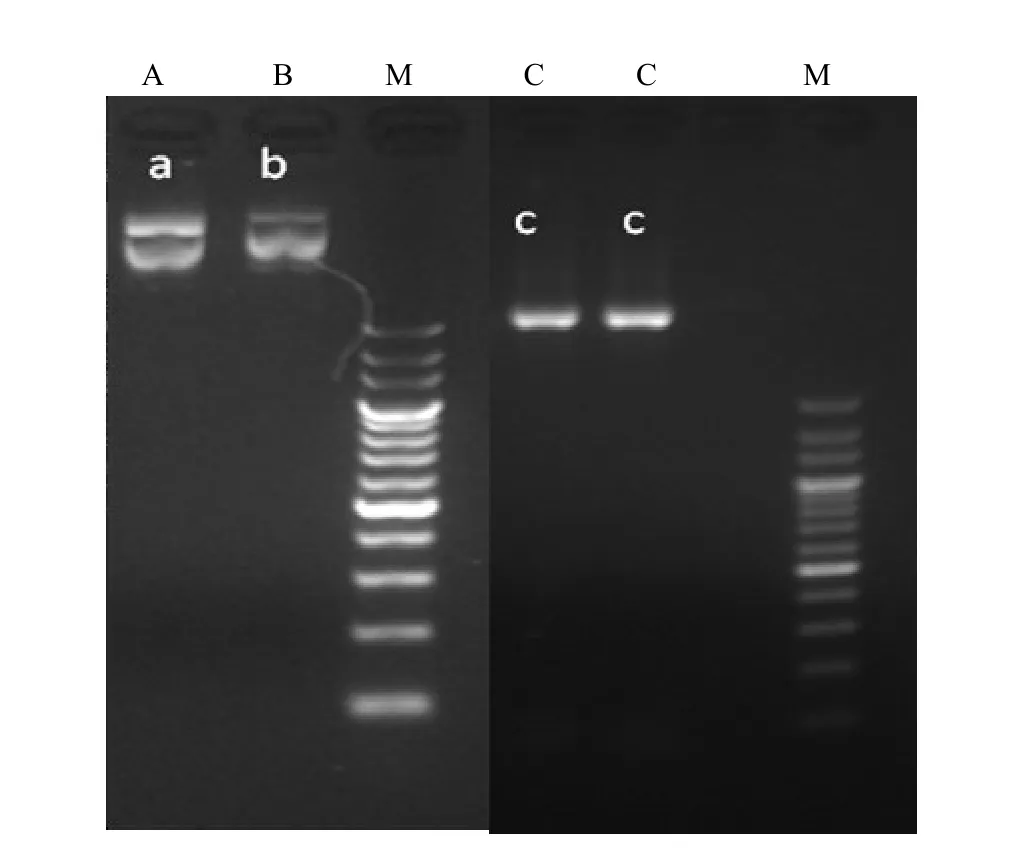
注:A:pYLgRNA-AtU3d-LacZ,B:pYLgRNA-AtU3b,C:pYLCRISPR/Cas9P35S-H,M:marker。Note:A:pYLgRNA-AtU3d-LacZ,B:pYLgRNA-AtU3b,C:pYLCRISPR/Cas9P35S-H,M:marker.图3 质粒电泳图Fig.3 The electrophoresis analysis of plasmids
2.3sgRNA表达盒的构建
sgRNA需要在植物细胞内合成从而指导Cas9蛋白编辑靶基因,而sgRNA的合成是通过sgRNA表达盒来完成,sgRNA表达盒由启动子序列和gRNA序列组成。pYLgRNA载体(pYLgRNA-AtU3d-LacZ、pYLgRNA-AtU3b)提供了不同的snRNA启动子可在植物细胞核内转录gRNA,并使gRNA停留在细胞核内发挥其功能。此外,pYLgRNA载体还提供gRNA中保守序列。因此,以pYLgRNA质粒为模板,通过常规PCR可以获得snRNA启动子序列和gRNA中保守序列,而gRNA中5′段的种子序列在引物合成时导入,联合应用常规PCR和overlapping PCR将启动子序列和gRNA序列组装在一起(图4)。电泳分析表明(图5),针对靶序列 T1的第一轮PCR获得了 360 bp 和130 bp左右的两个片段,分别为U3d启动子和连接T1靶序列的gRNA;而针对靶序列T2的第一轮PCR获得了380 bp和130 bp左右的两个片段,分别为U3b启动子和连接T1靶序列的gRNA;针对靶序列T1的第二轮PCR 将U3d启动子片段和gRNA片段2个分离的片段连接在一起组装成T1sgRNA表达盒;针对靶序列T2的第二轮PCR将U3b启动子片段和gRNA片段2个分离的片段连接在一起组装成T2sgRNA表达盒。
2.4两个sgRNA表达盒的拼接及与Cas9双元表达载体的组合
T1sgRNA和T2sgRNA两个单独的表达盒构建好后,需要将两个表达盒拼接在一起,并且插入Cas9双元表达载体中,以便通过农杆菌的介导整合到茶树基因组中,从而在茶树细胞内进行表达,获得Cas9和gRNA,进而对目标基因进行编辑。我们应用 Golden gate cloning 技术,一步完成sgRNA表达盒的拼接及与 Cas9双元表达载体的组合,从而获得CRISPR/Cas编辑载体(图6)。如图7所示,酶切、连接产物可见一条 980 bp左右的电泳条带,表明T1sgRNA和T2sgRNA表达盒连接成功,同时还可见一条>4 000 bp的电泳条带,说明T1sgRNA和T2sgRNA表达盒可能已组装进入 pYLCRISPR/Cas9P35S-H双元表达载体。连接产物转化 DH5α后摇菌提取质粒,以Mlu I酶切,结果如图所示,可见两条电泳条带,其中一条 950 bp左右的条带为T1sgRNA和T2sgRNA连接片段,而pYLCRISPR/Cas9P35S-H双元表达载体未见950 bp左右的电泳条带,说明酶切、连接成功,获得了TCS CRISPR/Cas9基因编辑载体。
2.5测序验证
为了进一步验证TCS 的CRISPR/Cas9基因编辑载体是否构建成功,将获得的10个质粒送上海生物工程技术有限公司测序,测序引物使用 SP-L2和 SP-R-C。测序结果(图 8)表明,T1sgRNA和T2sgRNA表达盒构建成功,在119 bp处检测到T1、T2靶序列,紧接靶序列后检测到 gRNA保守序列的存在,同时在T1和T2靶序列上游分别检测到U3d和U3b启动子序列,在 T1sgRNA 表达盒上游T2sgRNA表达盒下游均检测到了pYLCRISPR/Cas9P35S-H双元表达载体序列。测序结果(图 9)说明,T1sgRNA表达盒和T2sgRNA 表达盒构建成功,并成功组装入pYLCRISPR/Cas9P35S-H双元表达载体,证实TCS的CRISPR/Cas9基因编辑载体构建成功。

图4 sgRNA表达盒的构建流程Fig.4 Procedures for generation of a sgRNA expression cassette

图5 第一、二轮PCR反应产物电泳图Fig.5 The electrophoresis analysis of the first and the second round of PCR products
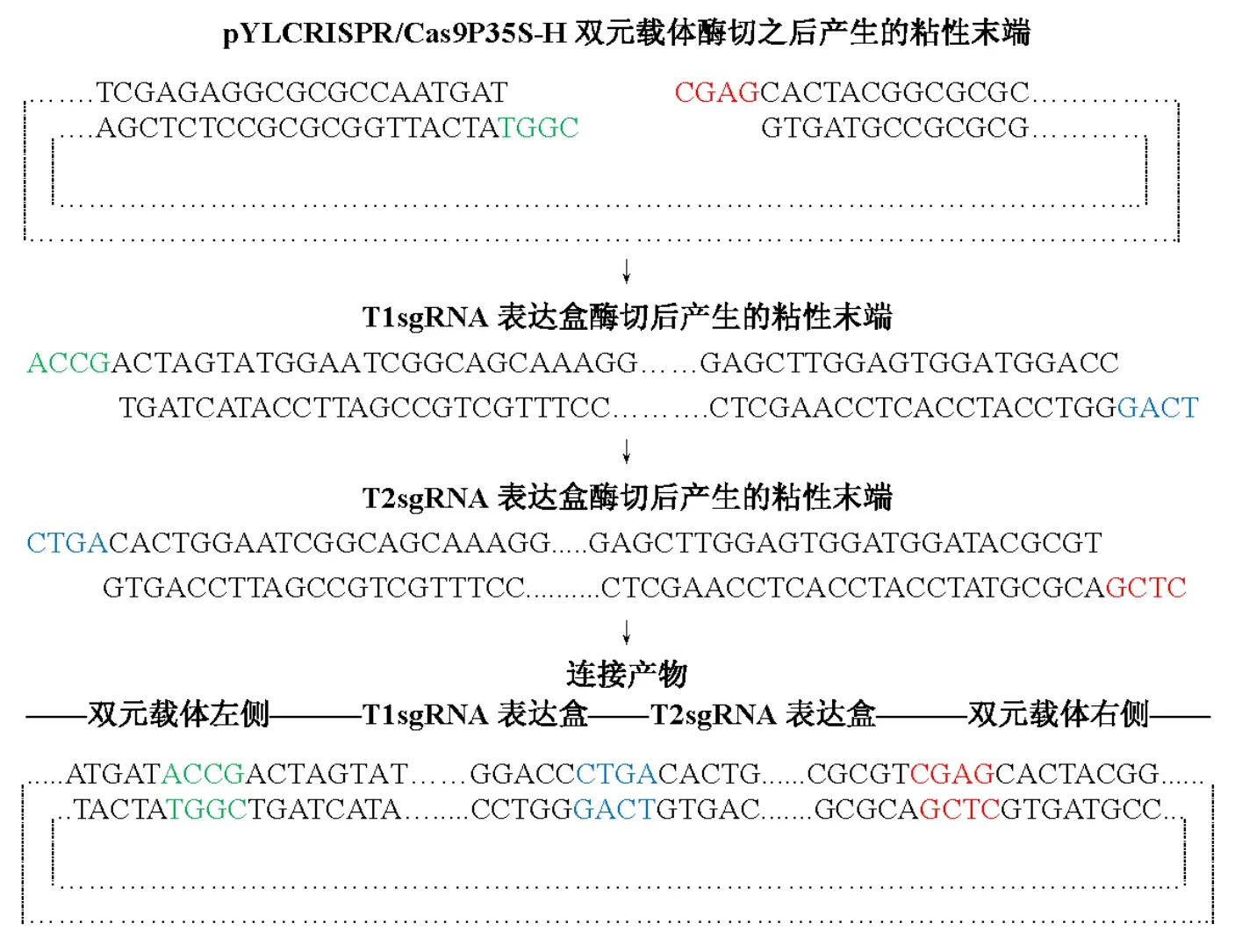
图6 Golden gate cloning 组装茶树CRISPR/Cas9基因组编辑载体Fig.6 Asembly the CRISPR/Cas9 editing vector by using golden gate cloning.

注:M为maker;L为sgRNA表达盒与Cas9双元表达载体酶切、连接产物;TCA为CRISPR/Cas9基因编辑载体的Mlu I酶切产物;C为质粒pYLCRISPR/Cas9P35S-H的Mlu I酶切产物。Note:M:maker.L:Digestion and ligation products of sgRNAs and pYLCRISPR/Cas9P35S-H.TCA:Digestion products of CRISPR/Cas9 cassettes for TCA with Mlu I.C:Digestion products of pYLCRISPR/Cas9P35S-H with Mlu I.图7 连接产物及质粒酶切电泳图Fig.7 The electrophoresis analysis of ligation and digestion products

图8 TCS基因编辑载体部分测序图Fig.8 The sequencing chromatogram of genome editing vector for TCS

图9 TCS基因编辑载体测序结果Fig.9 The sequence of genome editing vector for TCS
3 讨论
CRISPR是一种简单的,在全基因组水平上选择性调控目的基因表达的方法。该技术不同于传统的RNA干扰技术(RNAi),CRISPR干扰可以同时沉默任意数量的单个基因。此外,它能够更明确、更稳定地发挥作用,能够极其灵活和快速选择靶位点。2013年,张峰等[14]首先利用 CRISPR/Cas9技术实现了真核生物基因组的剪切。此后,CRISPR/Cas9技术研究一发不可收拾,国内外学者迅速在多种动、植物中开展了CRISPR/Cas9研究,不到3年的时间就已经在模式植物、农作物及园艺植物中取得了非常成功的应用[17]。据报道,在拟南芥和水稻中,CRISPR/Cas9系统引起的突变率高达90%以上。通常情况下,CRISPR/Cas9会产生野生株、杂合突变体、双等位基因突变体和纯合突变体,而只有双等位基因突变体和纯合突变体才有应用价值。因此,如果第一代转基因未能获得双等位基因突变体或纯合突变体需要通过不断自交从子代中筛选双等位基因突变体和纯合突变体。幸运的是,业已证明多种植物均能在第一代转基因中获得双等位基因突变体或纯合突变体,从而增加了 CRISPR/Cas9实用性。因此,对于茶树等自交不亲和的作物同样可以采用CRISPR/Cas9技术获得纯合突变株。但是,将CRISPR/Cas9技术应用于茶树中将面临表达载体构建的问题,目前尚无相关报道。本文以茶树中咖啡碱合成酶为例,联合采用常规PCR、Overlapping PCR和Golden Gate Cloning技术,构建了包含茶树咖啡碱合成酶双靶点的CRISPR/Cas9基因编辑载体,为CRISPR/Cas9基因编辑技术在茶树中的应用奠定了理论基础,同时对其他植物 CRISPR/Cas9基因编辑载体的构建提供了技术支撑。
虽然茶树基因组尚未释放,影响脱靶效应分析。但是,一般来说 CRISPR/Cas9要对基因编码区进行编辑才能引起突变表型,在没有全基因组序列的情况下对转录组序列进行比对,可在一定程度上排除错靶现象。此外,相对动物而言,植物的脱靶带来的风险较小。所以,在茶树基因组释放前,同样可以开展针对茶树的CRISPR/Cas9研究工作。
本文建立的 CRISPR/Cas9基因组编辑载体的构建方法,在Ma等[28]报道的方法上进行了完善与改进。pYLCRISPR/Cas9P35S-H等双元表达载体因属于低拷贝载体,且载体较大,一般需要采用大提质粒试剂盒来提取,具有成本高、效率低的缺点。本文通过扩大菌的培养量和培养时间,菌液约5 mL左右,震荡培养约 16 h,洗脱质粒时使用 60℃无菌水,且在烘箱中60℃孵育2 min,最后发现,采用小提质粒提取试剂盒同样可以获得高质量且足量的pYLCRISPR/Cas9P35S-H质粒(图3),完全满足载体构建要求。
第一轮PCR的目的是将gRNA及其启动子分别克隆出来,而第二轮 PCR是利用Overlapping PCR将和启动子连接起来形成gRNA表达盒,并在gRNA表达盒两端加上带BsaI酶切位点的接头,然后酶切连接,将各个gRNA表达盒连接起来,并与pYLCRISPRcas9载体连接。在第一轮PCRMa等[28]人将第一个反应与第2个反应在同一PCR管中进行,在一个反应中使用4种引物,在循环初始阶段同时扩增两个片段,并在循环后阶段通过Overlapping PCR将两片段拼接在一起。然而,我们实验结果表明,同一PCR管中进行2个反应很难获得特异片断。最后,我们将2个反应分开进行,获得了理想的结果。
本载体系统采用 Golden Gate Cloning[29]技术组装Cas9载体和多个gRNA表达盒片段。Golden Gate cloning是利用 BsaI(type IIs restriction enzyme)的识别位点和切割位点不重叠的特性,可以设计出多种不同的且非回文结构的粘性末端(256-16=240种,减去的 16种是回文结构)。多个要连接的片段在连接点具有特异的互补末端可以连接,而非连接点的末端之间不互补不能连接(相同末端分子间也不互补不能连接),因此连接反应是向着目标单方向进行,效率很高。此外,利用 BsaI酶进行Golden Gate cloning时,可实现酶切、连接同步化,即边切边连,不需要酶切后切胶回收的繁琐操作,简单、方便且节省大量时间。Ma等[28]采用的酶切、连接程序为:10℃ 5 min,20℃ 5 min;10~15循环。按照Ma等[28]等方法我们尝试多次,但均未成功,后来对反应条件进行了多次优化,最后确定了酶切、连接程序为:37℃ 1 min,16℃ 1 min,30个循环,最后55℃灭活内切酶5 min。
本文成功构建了靶向TCS的CRISPR/Cas9基因组编辑载体,为茶树其他基因靶向的 CRISPR/Cas9基因组编辑载体的构建提供了借鉴方法。下一步的工作,是通过根癌农杆菌的介导将 CRISPR/Cas9基因组编辑载体转入茶叶愈伤组织,获得转基因愈伤组织或转基因茶树,并检测突变材料中的咖啡碱生物合成情况,验证 CRISPR/Cas9基因组编辑载体的有效性。如有必要,再对载体进行改进,比如使用茶树本身的 sn RNA启动子驱动gRNA的表达,根据茶树的特性优化 Cas9密码子,从而建立完善的 CRISPR/Cas9介导的茶树基因组编辑体系,推动茶树分子育种、基因功能研究、茶树基因工程等工作快速发展。
[1]Shi CY,Yang H,Wei CL,et al.Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds [J].BMC Genomics,2011,12(1):1-19.
[2]Liu SC,Jin JQ,Ma JQ,et al.Transcriptomic analysis of tea plant responding to drought stress and recovery [J].PLoS ONE,2016,11(1):e0147306.http://dx.doi.org/10.1371/journal.pone.0147306.
[3]Wei Y,Jing W,Youxiang Z,et al.Genome-wide identification of genes probably relevant to the uniqueness of tea plant(Camellia sinensis)and its cultivars [J].International Journal of Genomics,2015,2015:527054.doi:10.1155/2015/527054.
[4]Wang XC,Zhao QY,Ma CL,et al.Global transcriptome profiles of Camellia sinensis during cold acclimation [J].BMC Genomics 2013,14:415.DOI:10.1186/1471-2164-14-415.
[5]Mukhopadhyay M,Mondal TK,Chand PK.Biotechnological advances in tea(Camellia sinensis [L.]O.Kuntze):a review [J].Plant Cell Reports,2015,35(2):255-287.
[6]Fauser F,Roth N,Pacher M,et al.In planta gene targeting [J].Proc Natl Acad Sci USA,2012,109(19):7535-7540.
[7]Li T,Liu B,Spalding MH,et al.High-efficiency TALEN-based gene editing produces disease-resistant rice [J].Nat Biotech,2012,30(5):390-392.
[8]Belhaj K,Chaparro-Garcia A,Kamoun S,et al.Editing plant genomes with CRISPR/Cas9 [J].Current Opinion in Biotechnology,2015,32:76-84.
[9]Mali P,Yang LH,Esvelt KM,et al.RNA-Guided human genome engineering via Cas9 [J].Science,2013,339(6121):823-826.
[10]Xie K,Yang Y.RNA-guided genome editing in plants using a CRISPR-Cas system [J].Mol Plant,2013,6(6):1975-1983.[11]Schaeffer SM,Nakata PA.CRISPR/Cas9-mediated genome editing and gene replacement in plants:transitioning from lab to field [J].Plant Sci,2015,240:130-142.
[12]Wiedenheft B,Sternberg SH,Doudna JA.RNA-guided genetic silencing systems in bacteria and archaea [J].Nature,2012,482(7385):331-338.
[13]Jinek M,Chylinski K,Fonfara I,et al.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity [J].Science,2012,337(6096):816-821.
[14]Wang HY,Yang H,Shivalila CS,et al.One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering [J].Cell,2013,153(4):910-918.
[15]Schiml S,Puchta H.Revolutionizing plant biology:multiple ways of genome engineering by CRISPR/Cas [J].Plant Methods,2016,12(1):1-9.
[16]Lintner NG,Frankel KA,Tsutakawa SE,et al.The structure of the crispr-associated protein csa3 provides insight into the regulation of the CRISPR/Cas system [J].Journal of Molecular Biology,2011,405(4):939-955.
[17]Gilbert LA,Larson MH,Morsut L,et al.CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes [J].Cell,2013,154(2):442-451.
[18]Nekrasov V,Staskawicz B,Weigel D,et al.Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease [J].Nat Biotech,2013,31(8):691-693.
[19]Jiang WZ,Zhou HB,Bi HH,et al.Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis,tobacco,sorghum and rice [J].Nucleic Acids Res,2013,41(20):e188.
[20]Wang Z,Xing H,Dong L,et al.Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generateshomozygous mutants for multiple target genes in Arabidopsis in a single generation [J].Genome Biol,2015,16:144.
[21]Upadhyay SJ,Alok A,Tuli R.RNA-guided genome editing for target gene mutations in wheat [J].G3(Bethesda).2013,3(12):2233-2238.DOI:10.1534/g3.113.008847.
[22]Liang Z,Zhang K,Chen KL,et al.Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system [J].J Genetics Genomics,2014,41(2):63-68.
[23]Zhang H,Zhang J,Wei P,et al.The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation [J].Plant Biotechnol J,2014,12(6):797-807.
[24]Čermák T,Baltes NJ,Čegan R,et al.High-frequency,precise modification of the tomato genome [J].Genome Biol,
[25]Jia H,Wang N.Targeted genome editing of sweet orange using Cas9/sgRNA [J].PLoS ONE,2014,9(4):e93806.DOI:10.1371/journal.pone.0093806.
[26]Fan D,Liu T,Li C,et al.Efficient CRISPR/Cas9-mediated targeted mutagenesis in populus in the first generation [J].Scientific Reports,2015,5:12217.
[27]Zhang B,Yang X,Yang C,et al.Exploiting the CRISPR/Cas9 system for targeted genome mutagenesis in petunia [J].Scientific Reports,2016,6:20315.
[28]Ma X,Zhang Q,Zhu Q,et al.A Robust CRISPR/Cas9 system for convenient,high-efficiency multiplex genome editing in monocot and dicot plants [J].Mol Plant,2015,8(8):1274-1284.
[29]Marillonnet S,Werner S.Assembly of multigene constructs using golden gate cloning [J].Methods Mol Biol,2015,1321:269-284.
Development of a CRISPR/Cas9 Constructed for Genome Editing of Caffeine Synthase in Camellia sinensis
TANG Yuwei1,3,LIU Liping1,3,WANG Ruoxian1,CHEN Yuhong1,LIU Zhonghua1,2,3,LIU Shuoqian1,2,3*
1.College of Horticulture and Hardening,Hunan Agricultural University,Changsha 410128,China;
2.National Research Center of Engineering Technology for Utilization of Functional Ingredients from Botanicals,Changsha 410128,China;
3.Key Lab of Tea Science,Ministry of Education,Changsha 410128,China
CRISPR/Cas9 technology(clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9)is a novel and powerful approach for targeted genome editing,such as targeted gene knock out or site-directed mutagenesis in a simple and easy way.Since its establishment,the CRISPR/Cas9 technique has been successfully applied in many eukaryotic organisms,including more than 10 plant species.However,it has not been available for genome editing of tea plant [Camellia sinensis(L.)O.Kuntze]due to the difficulty in constructing CRISPR/Cas9 expression vector.The present work developed an efficient method to construct a CRISPR/Cas9 expression vector for genome editing a tea caffeine synthase(TCS)by using general PCR,overlapping PCR and golden gate cloning technology.The present work would promote the application of CRISPR/Cas9 technology in genomic modification in tea plants.
Camellia sinensis(L.),genome editing technology,tea caffeine synthase,CRISPR/Cas9 technique
TS272;Q52
A
1000-369X(2016)04-414-13
2016-04-25
2016-05-12
唐雨薇,女,硕士研究生,主要从事茶树分子生物学研究,*通讯作者:shqianliu@sina.com
2015,16:232.10.1186/s13059-015-0796-9.
