基于快速和常规全外显子组分析技术对遗传性疾病的诊断程序比较
2016-09-19魏泽峻卢宇蓝吴冰冰王慧君周文浩
杨 琳 董 辰 魏泽峻 卢宇蓝 吴冰冰 王慧君 周文浩
·论著·
基于快速和常规全外显子组分析技术对遗传性疾病的诊断程序比较
杨琳1,3董辰1,3魏泽峻2卢宇蓝1吴冰冰1王慧君1周文浩1
目的探索可被临床医生直接应用的全外显子测序(WES)数据分析平台及方法。方法选择复旦大学附属儿科医院(我院)临床诊断不明疾病的核心家系3例(例1~3)和仅先证者2例(例4和5)行WES分析,采用WuXi NextCODE临床测序数据分析系统(简称NextCODE平台)进行快速数据分析,并与我院分子诊断中心已建立的高通量数据分析流程(简称常规数据分析流程)进行参与人员及耗时的比较。结果例1~3基于表型相关候选基因联合遗传模式的分析方法,分别检测到FGFR2基因杂合突变、GBE1基因复合杂合突变及TBX1基因杂合突变;例4和5通过表型相关候选基因分析,分别检测到IL10RA基因纯合突变和复合杂合突变。NextCODE平台自动完成3/7个步骤,从输入表型至生成报告,WES数据分析用时30 min以内。常规数据分析流程自动完成1/7个步骤,6个人工完成步骤需要多个专业人员进行数据的筛选及解读,从输入表型至生成报告,熟练的专业人员用时2~8 h。结论5例临床诊断不明病例通过WES明确了诊断;NextCODE是直接为临床医生所用、简单快速的WES数据分析平台,有助于协助临床医生直接利用高通量测序数据,准确锁定致病突变,提高诊断效率。
全外显子测序;诊断不明病例;WuXi NextCODE数据分析系统
截至2016年4月15日人类孟德尔遗传病数据库中[1],明确遗传学病因的疾病达4 705种。高通量测序技术可以在短时间内获得大量遗传信息,在快速寻找致病突变、明确诊断、精准治疗和产前咨询等方面有广阔前景。外显子组即一个个体的基因组DNA上所有蛋白质编码序列 (外显子)的总和。预计85%的人类致病突变都位于这1%的蛋白质编码序列上[2]。在过去的3年中,将全外显子测序(WES)用于儿科临床诊断不明的综合征已经取得了很多的进展 ,至少有150种遗传性疾病发现了新的致病基因,或将已知致病基因与新的表型建立了关联[3]。通过WES获得的海量遗传变异信息,从中挖掘出有效信息,需要临床遗传分析平台具有计算机、生物信息和临床遗传学等多方面的能力[4~8]。为更广大的临床医生较为便利地应用高通量数据分析病例,解释临床现象。WuXi NextCODE临床测序数据分析平台(简称NextCODE平台),通过表型-遗传模式综合筛选、致病突变筛选、结合多数据库资源整合进行快速数据分析,使临床医生无需有专门的生物信息知识和硬件设备,就可以对高通量测序数据进行方便、快捷、准确地读取和分析。
本文对诊断不明的儿科遗传性疾病,以NextCODE平台进行快速数据分析,并与复旦大学附属儿科医院(我院)分子诊断中心已经建立的高通量数据分析流程[7](简称常规数据分析流程)进行参与人员和耗时的比较。
1 方法
1.1分析病例选择选取我院临床诊断不明的核心家系和仅先证者病例。本研究经我院伦理委员会批准。
1.2DNA提取及全外显子组捕获采用QIAGEN公司mini blood全血试剂盒及其标准DNA抽提方法提取基因组DNA(gDNA),用美国Thermofisher公司生产的NanoDrop紫外分光光度仪测定样本的浓度及定量。参照SureSelct Human All Exon试剂盒说明书,基因组DNA经过超声打断、末端修复、接头连接和杂交捕获。捕获文库采用Illumina HiSeq2000平台进行序列检测。原始图像文件经Illumina base calling Software 1.7进行图像识别(Base calling),去除污染及接头序列处理后。Clean reads采用Burrows-Wheeler Aligner(BWA)软件v.0.5.9-r16,以人类基因组hg19(GRCh37)为参考序列进行比对。
1.3NextCODE平台数据分析系统
1.3.1输入数据①常规高通量测序生成的BAM文件,即测序所得片段在基因组上的比对信息;②提供基因组上突变信息的VCF文件,包括SNV和INDEL;③先证者和(或)核心家系的临床信息。
1.3.2NextCODE平台的序列分析工具(Clinical Sequence Analyzer, CSA)系统①基因组信息注释,采用Ensembl数据库(http://www.ensembl.org/index.html)和RefSeq(The Reference Sequence, http://www.ncbi.nlm.nih.gov/refseq/);②对基因/变异与疾病关系注释,采用OMIM、HGMD(http://www.hgmd.cf.ac.uk/ac/index.php)和ClinVar(http://www.nlm.nih.gov/clinvar)数据库;③突变位点的相关疾病注释,采用临床基因组数据库CGD(http://research.nhgri.nih.gov/CGD)和EuroGentest项目数据库(www.eurogentest.org);④突变功能注释,采用VEP工具(http://asia.ensembl.org/info/docs/tools/vep/index.html);⑤注释变异频率,采用千人基因组计划(1000 Genomes, http://www.1000genomes.org)、NHLBI外显子组测序项目(NHLBI-ESP,http://evs.gs.washington.edu/EVS/)和The Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/);⑥突变位点的致病性评级,根据美国遗传学学会(ACMG)突变解读标准指南。
NextCODE平台用于测序数据深度解读的Sequence Miner(SM),可以可视化查看突变位点测序片段匹配情况。直接点击突变的位置即可跳转至该突变位点区域每个测序片段在基因组上的匹配情况、区域覆盖深度和测序质量等。
1.4常规WES数据分析参见文献[7],以我院分子诊断中心有500例以上的WES数据分析经历的熟练专业人员为标准,估计用时。
1.5Sanger测序验证对于检测到的变异,采用Primer 3在线进行引物设计,使用KAPA 2G Robust Hot Start ReadMix进行扩增反应。通过Sanger直接测序法(3500XL Genetic Analyzer,ABI)进行验证,协助明确诊断。
2 结果

表15例先证者主要临床表型

编号家族史神经系统特殊面容循环/消化系统泌尿生殖/内分泌指趾/免疫功能/其他1,男 无头颅形态异常,CT示后颅窝结构拥挤前额突出,耳位低,眼距宽,高腭弓房间隔缺损/肝肿大B超提示肾脏结构不清双侧多指,双侧并指/趾2,男哥哥3月龄智力落后,1岁肝损害死亡肝肿大,肝功能异常3,女哥哥28周早产,出生体重500g,生后2d死亡哭声弱,头颅CT提示胼胝体压部可疑异常信号耳位低,耳廓贴颅,眼距宽,反复双眼向上凝视,鼻梁低平胃食管反流甲状旁腺功能减退CD3计数250,细胞免疫缺陷,低钙血症4,女生后2d出现惊厥反复腹泻,肛瘘,肛周脓肿,肠狭窄反复发热5,女G1P1流产,G2P2足月女婴,1月龄反复发热、腹泻,5月龄死亡腹泻,肛周脓肿,肛瘘,小肠狭窄PLT下降,发热
2.2NextCODE平台与常规数据分析流程参与人员及耗时的比较图1显示,NextCODE平台基本数据分析流程包括7个主要步骤:上传测序原始数据、选择样本建立研究、输入表型确定候选基因、选择遗传模式、与表型相关性分析、结果解读和生成报告;自动完成3个步骤;4个人工完成的步骤仅结果解读或可由临床遗传学医生协助,其余均可由临床医生完成;从输入表型至生成报告5个步骤,核心家系WES数据分析一般用时10~15 min,仅先证者WES数据分析一般用时30 min。
常规数据分析流程包括7个主要步骤:获得测序原始数据、拼接连接比对、获得变异结果、变异注释、生物信息学变异筛选、人工数据分析和报告书写;自动完成1个步骤;6个人工完成步骤需要多专业人员参与才能建立起高效可行的分析流程,并且需要熟练的数据分析人员,进行数据的筛选及解读;多专业人员包括生物信息、计算机专业和临床遗传学医生,其中3个步骤需要2个专业共同完成;从输入表型至生成报告5个步骤,无论是核心家系还是仅先证者WES数据分析基于熟练的专业人员用时2~8 h。

图1NextCODE平台和常规数据分析流程的WES数据分析
2.3基于NextCODE平台WES数据分析根据例1~5临床表现和输入不同的表型关键词。NextCODE平台WES数据分析结果如表2所示。
例1表型相关候选基因共757个;患儿为先证者,家族史中无特殊描述,父母均无异常表型,故支持新发突变可能性大,也可为常染色体隐性遗传(AR)模式;在候选基因检测到的变异中符合新发突变1个变异,为FGFR2基因NM_000141,c.940-2A>G;没有符合AR模式的变异。
例2表型相关候选基因共335个;患儿为先证者,其哥哥有相似表型,父母均无异常表型,故支持AR或X连锁遗传(XR)模式;由于患儿表型特异性不强,在表型相关候选基因中没有检测到符合AR或XR的变异;在CSA预置的panel里,其中儿童隐性遗传病panel检测到的变异中符合AR模式共2个变异,为GBE1基因NM_000158,c.C1402A:p.R468S和c.T964C:p.W322R,没有符合XR的变异。
例3表型相关候选基因共312个;患儿为先证者,家族史中其哥哥为28周早产死亡,父亲存在发音不清,母亲无异常表型,故支持常染色体显性遗传(AD);在候选基因检测到的变异中符合AD模式共8个变异,其中TBX1基因所导致疾病与患儿表型相符。
例4表型相关候选基因共1 028个;因没有家系数据可以进行遗传模式的选择,在表型相关候选基因检测到的35个变异中逐个根据已知的基因功能与疾病的关联性行手工分析,最终锁定IL10RA基因纯合突变:NM_001558,c.C301T:p.R101W。
例5表型相关候选基因共356个;因没有家系数据可以进行遗传模式的选择,在表型相关候选基因检测到的14个变异中逐个根据已知的基因功能、与疾病的关联性行手工分析,锁定IL10RA基因符合杂合突变NM_001558,c.T299G:p.V100G和c.C301T:p.R101W(图2)。
表2NextCODE平台WES数据分析结果
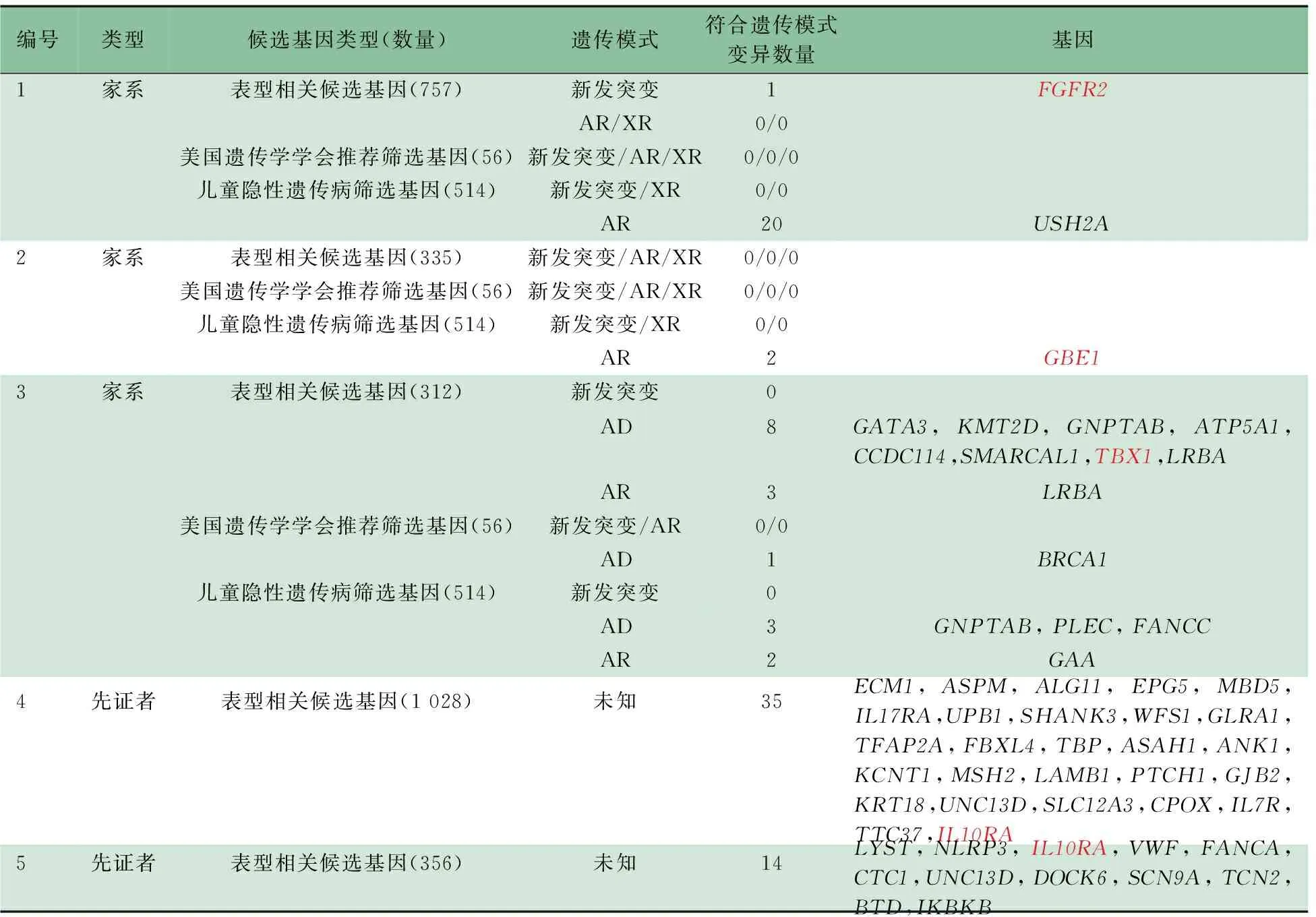
编号类型候选基因类型(数量)遗传模式符合遗传模式变异数量基因1家系表型相关候选基因(757)新发突变1FGFR2AR/XR0/0美国遗传学学会推荐筛选基因(56)新发突变/AR/XR0/0/0儿童隐性遗传病筛选基因(514)新发突变/XR0/0AR20USH2A2家系表型相关候选基因(335)新发突变/AR/XR0/0/0美国遗传学学会推荐筛选基因(56)新发突变/AR/XR0/0/0儿童隐性遗传病筛选基因(514)新发突变/XR0/0AR2GBE13家系表型相关候选基因(312)新发突变0AD8GATA3,KMT2D,GNPTAB,ATP5A1,CCDC114,SMARCAL1,TBX1,LRBAAR3LRBA美国遗传学学会推荐筛选基因(56)新发突变/AR0/0AD1BRCA1儿童隐性遗传病筛选基因(514)新发突变0AD3GNPTAB,PLEC,FANCCAR2GAA4先证者表型相关候选基因(1028)未知35ECM1,ASPM,ALG11,EPG5,MBD5,IL17RA,UPB1,SHANK3,WFS1,GLRA1,TFAP2A,FBXL4,TBP,ASAH1,ANK1,KCNT1,MSH2,LAMB1,PTCH1,GJB2,KRT18,UNC13D,SLC12A3,CPOX,IL7R,TTC37,IL10RA5先证者表型相关候选基因(356)未知14LYST,NLRP3,IL10RA,VWF,FANCA,CTC1,UNC13D,DOCK6,SCN9A,TCN2,BTD,IKBKB
注AR: 常染色体隐性遗传; AD:常染色体显性遗传; XR: X连锁隐性遗传;基因中红色字体为致病突变所在基因

图2Sequence Miner显示先证者(例5)检测到的IL10RA基因突变位点图
注A:红色柱状显示突变位点所在区域在染色体上的定位;B:突变位点所在区域正反向碱基的参考排列顺序;C:测序产生的reads与参考序列比对的结果:其中橘黄色和蓝色的reads分别代表正反2个方向;黄色突出的碱基为突变的碱基;蓝色竖线指示的突变位点参考序列为T,部分reads上突变为G,即c.T299G:p.V100G位点杂合突变;位于其后间隔1个碱基的位置参考序列为C,部分reads上突变为T即c.C301T:p.R101W杂合突变
3 讨论
3.1NextCODE平台对于家系遗传疾病的WES数据分析本文分析例1~3为家系WES数据,采用NextCODE平台CSA系统进行分析时,通过表型缩小候选基因范围,其中例1和3,致病突变均在表型相关基因中。再通过家系遗传模式的进一步删选,直接锁定致病突变,整个分析过程简单、快捷和准确。其中,例1检测到得FGFR2基因的剪切位点突变为已知Pfeiffer综合征致病位点[9,10],Pfeiffer综合征为AD疾病,主要表现为颅缝早闭、面中部发育不良和并指趾畸形[11],与患儿表型相符,支持Pfeiffer综合征诊断。例3符合AD模式的共8个变异,分别为GATA3、KMT2D、GNPTAB、ATP5A1、CCDC114、SMARCAL1、TBX1和LRBA基因,其中TBX1基因NM_080647,c.G385A:p.E129K,既往针对非22q11缺失的圆锥动脉干畸形患儿检出该位点的突变[12]。例3特殊面容、细胞免疫功能缺陷、胸腺缺如及低钙血症,临床高度怀疑为22q11微缺失所致[13],但是细胞遗传学检测没有发现该区段的异常,行WES检测,且检测到得TBX1基因突变符合临床表型[14],明确诊断。
CSA系统内置了3组已知致病基因,一是ACMG推荐基因,包括56个在临床外显子和全基因组测序检测诊断中需要报道的致病基因[15],覆盖24种疾病。儿童隐性遗传病基因,包含由Stephen Kingsmore总结的514个存在已知致病突变的基因。这些基因在儿童遗传病诊断中被建议优先考虑[16]。线粒体疾病基因(mitochondria),包含2 659个对线粒体功能有影响的染色体/线粒体基因。此外,CSA高级分析也支持加入自定义候选基因作为筛选条件。除了候选基因分析模式中的3组基因以外,CSA在高级分析中也内置了几组其他类型致病基因可供选择,包括致癌基因(cancer,87个基因)、先天性心脏病基因(cardiomyopathy,41个基因)、纤毛疾病基因(cillia,2 737个基因)、免疫系统疾病(immuno,910个基因)和美国国立癌症研究所推荐的遗传性癌症致病基因(nci37,35个)。
例2表型中仅有肝肿大、肝功能异常的非特征性表型,没有在表型相关候选基因中找到致病突变,在儿童常见隐性遗传病panel中,检测到了GBE1基因的复合杂合突变c.C1402A:p.R468S和c.T964C:p.W322R。GBE1基因为糖原累积症Ⅳ的致病基因[17],可以解释患儿及其已经死亡的哥哥的表型。
总之,NextCODE平台对于家系WES数据,基于表型相关候选基因联合遗传模式的分析方法,可以减小数据分析的压力,减少手工分析的环节,缩短数据分析的时间,较快的形成诊断。
3.2NextCODE平台对于先证者遗传疾病的WES数据分析本文例4和5尚未发现家系原因所致疾病。主要表型均为腹泻、肛瘘、肛周脓肿及肠狭窄,例4还有惊厥表型,故表型相关候选基因远多于例5。根据表型相关候选基因检测到的变异行逐个人工筛选,均锁定IL10RA基因,导致早发的炎症性肠病,符合AR模式[18]。其中例4为R101W纯合突变,该位点为炎症性肠病已知的致病位点[19]。例5为R101W和V100G复合杂合突变。
例2和5为复合杂合突变,在没有进行父母验证之前,需要通过原始数据协助判断2个突变非同一来源。NextCODE平台提供的SM分析工具,可以查看突变位点测序片段匹配情况。如突变位点区域每个测序片段在基因组上的匹配情况以及该区域的覆盖深度和测序质量等(图2),对于家系样本,可以同时看到突变位点在不同成员中的分布情况。此外,SM还提供了不同数据库在区域的注释信息,例如Ensembl、ClinVar、UCSC Genome Browser和dbSNP等,可以方便进行相关数据库的比较。
3.3NextCODE平台数据分析参与人员NextCODE平台自动完成步骤比例较高,且大部分手工步骤可由临床医生自行完成,仅结果解读需要临床遗传学专科医生的协助。
常规数据分析流程,需要多专业人员共同参与,才能建立起高效可行的分析流程,并且需要熟练的数据分析人员,进行数据的筛选及解读。但是,对于临床表型不详细或不典型,不能直接锁定致病突变的病例,采用常规WES数据分析流程,可以对于每个变异进行详细的评估及注释标记;可以根据每个病例的特殊性进行细微的调整。对于熟练的专业数据分析人员,也可以较快的完成WES数据分析。
随着测序成本的不断降低和临床对于高通量测序认识的不断深入,一方面,临床不能诊断的疾病、有效/敏感药物的选择、遗传相关危象的快速筛查等,越来越依赖于先进的分子诊断技术,另一方面,将需要经过长期专业培训才能使用的WES数据分析流程,转化为可以直接为临床医生所使用的平台,对临床同样是必要的和现实的。WES数据分析流程的智能化,使得医生无需借助生物信息平台和相关专业人员处理高通量测序数据,即可找到准确和可靠的致病突变,提高诊断效率,是大势所趋。
致谢感谢患儿及其家属的积极参与和配合;感谢明码(上海)生物科技有限公司提供的支持和帮助。
[1] Schorderet DF.Using OMIM (On-line Mendelian Inheritance in Man) as an expert system in medical genetics.Am J Med Genet,1991,39 (3):278-284
[2] Choi M,Scholl UI,Ji W,et al.Genetic diagnosis by whole exome capture and massively parallel DNA sequencing.Proc Natl Acad Sci U S A,2009,106 (45):19096-19101
[3] Grody WW,Thompson BH,Hudgins L.Whole-exome /genome sequencing and genomics.Pediatrics,2013,132 (S3):211-215
[4] Need AC,Shashi V,Hitomi Y,et al.Clinical application of exome sequencing in undiagnosed genetic conditions.J Med Genet,2012,49(6):353-361
[5] Hane Lee P,Joshua L.Deignan P,Naghmeh Dorrani M,CGC,et al.Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders.JAMA,2014,312(18):1880-1887
[6] Frederick E.Dewey M,Megan E.Grove M,Cuiping Pan P,et al.Clinical Interpretation and Implications of Whole-Genome Sequencing.JAMA,2014,311 (10):1035-1044
[7]黎籽秀,刘博,徐凌丽,等.高通量测序数据分析和临床诊断流程的解读.中国循证儿科杂志,2015,10 (1):19-24中国循证儿科杂志2016 年4 月第11 卷第2 期·135·
[8]Yang Y,Muzny DM,Reid JG,et al.Clinical whole-exome sequencing for the diagnosis of mendelian disorders.N Engl J Med,2013,369 (16):1502-1511
[9]Jang JH,Shin KH,Park JG.Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers.Cancer Res,2001,61 (9):3541-3543
[10]Schell U,Hehr A,Feldman GJ,et al.Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome.Hum Mol Genet,1995,4 (3):323-328
[11]Plomp AS,Hamel BC,Cobben JM,et al.Pfeiffer syndrome type 2:further delineation and review of the literature.Am J Med Genet,1998,75 (3):245-251
[12]Xu YJ,Chen S,Zhang J,et al.Novel TBX1 loss-of-function mutation causes isolated conotruncal heart defects in Chinese patients without 22q11.2 deletion.BMC Med Genet,2014,15:78
[13]Bartsch O,Nemeckova M,Kocarek E,et al.DiGeorge /velocardiofacial syndrome:FISH studies of chromosomes 22q11 and 10p14,and clinical reports on the proximal 22q11 deletion.Am J Med Genet A,2003,117A (1):1-5
[14]Yagi H,Furutani Y,Hamada H,et al.Role of TBX1 in human del22q11.2 syndrome.Lancet,2003,362 (9393):1366-1373
[15]Green RC,Berg JS,Grody WW,et al.ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing.Genet Med,2013,15 (7): 565-574
[16]Kingsmore S.Comprehensive carrier screening and molecular diagnostic testing for recessive childhood diseases.PLoS Curr,2012,e4f9877ab8ffa9
[17]Bao Y,Kishnani P,Wu JY,et al.Hepatic and neuromuscular forms of glycogen storage disease type IV causedby mutations in the same glycogen-branching enzyme gene.J Clin Invest,1996,97 (4):941-948
[18]Gasche C,Grundtner P,Zwirn P,et al.Novel variants of the IL-10 receptor 1 affect inhibition of monocyte TNF-alpha production.J Immunol,2003,170(11):5578-5582
[19]Mao H,Yang W,Lee PP,et al.Exome sequencing identifies novel compound heterozygous mutations of IL-10 receptor 1 in neonatal-onset Crohn's disease.Genes Immun,2012,13 (5):437-442
(本文编辑:张崇凡)
Procedure comparison between rapid and standard whole-exome data interpretation for clinical diagnosis of genetic disorder
YANGLin1,3,DONGChen1,3,WEIZe-jun2,LUYu-lan1,WUBin-bin1,WANGHui-jun1,ZHOUWen-hao1
(1Children′sHospitalofFudanUniversity,Shanghai201102; 2WuXiNextCODEGenorvics(Shanghai)Co;Ltd,Shanghai200131,China; 3Co-firstauthor)
ZHOU Wen-hao,E-mail:zwhchfu@126.com
Whole-exome sequencing; Undiagnosed cases;WuXi NextCODE software
10.3969/j.issn.1673-5501.2016.02.010
上海科学技术委员会/医学领域重点项目子课题:14411950402;上海市卫生和计划生育委员会课题:沪卫计科教[2013]018号
1 复旦大学附属儿科医院分子诊断中心上海,201102;2 明码(上海)生物科技有限公司上海,200131;3 共同第一作者
周文浩,E-mail:zwhchfu@126.com
2016-03-10
2016-04-03)
AbstractObjectiveA difficult hurdle in whole exome sequencing application is rapid data interpretation. In this study, whole-exome sequencing and WuXi NextCODE software were used to rapidly identify pathogenic mutations in 5 undiagnosed cases.MethodsThe exome targets of the patient′s DNA were captured with the SureSelct Human All Exon kit followed by sequencing with the Illumina HiSeq2000 platform. The WuXi NextCODE software was used for the data analysis. The detected variant was confirmed with Sanger direct sequencing. Results5 trio families with undiagnosed probands were recruited. A heterozygous missense mutation was identified inFGFR2 gene in proband 1, compound heterozygous missense mutations inGBE1 gene in proband 2, and a heterozygous missense mutation inTBX1 gene in proband 3 by whole-exome sequencing of trio samples. A homozygous missense mutation was identified inIL10RAgene in proband 4, and compound heterozygous missense mutations in IL10RA gene in proband 5 by whole-exome sequencing of proband only.ConclusionThis study clearly showed the efficacy of whole-exome sequencing and was helpful to rapid genetic diagnosis for undiagnosed cases.
