微乳液相色谱法测定厄贝沙坦片中厄贝沙坦的含量及有关物质
2016-09-14郑艾妮王柳云余诺君李宁广东药科大学药学院广东广州510006
郑艾妮,王柳云,余诺君,李宁(广东药科大学药学院,广东广州510006)
微乳液相色谱法测定厄贝沙坦片中厄贝沙坦的含量及有关物质
郑艾妮,王柳云,余诺君,李宁
(广东药科大学药学院,广东广州510006)
目的建立一种微乳液相色谱法测定厄贝沙坦片的质量分数及有关物质。方法 采用 Agela technologies odyssil C18柱(4.6 mm×250 mm,5 μm),以SDS-正丁醇-环己烷-水(质量比3.0∶6.0∶0.8∶92.2)为流动相,柱温30℃,流速1.0 mL/min,进样量20 μL,检测波长230 nm。结果有关物质与主要成分可以完全分离。厄贝沙坦在50.01~175.04 μg/mL范围内与峰面积线性关系良好(r=0.999 2,n=6),检测限为0.02 μg/mL,平均回收率为100.4%(RSD=1.0%,n=9)。有关物质在0.075 3~0.351 8 μg/mL范围内呈线性,检测限为0.025 μg/mL,平均回收率为100.5%(RSD=1.1%,n=9)。2个规格的厄贝沙坦片中厄贝沙坦的平均质量分数分别为99.4%和99.8%。结论建立的微乳液相色谱法检测厄贝沙坦及相关物质快速、准确、重复性好且绿色环保,减少了有机溶剂的使用。
厄贝沙坦片;微乳液相色谱法;厄贝沙坦;有关物质
厄贝沙坦是一种高选择性Ⅰ型血管紧张素Ⅱ受体拮抗剂,可用于治疗高血压[1],包括轻度至中度高血压[2]或2型糖尿病、肾病[3]。厄贝沙坦具有良好的降压作用,可通过阻断肾素-血管紧张素-醛固酮系统(RAS)达到控制血压。目前,厄贝沙坦的测定方法主要有HPLC/MS[4]、UPLC-MS/MS[5]、HPLC[5-10]和紫外分光光度法[11]。
厄贝沙坦是弱极性物质,中国药典和美国药典记载均使用磷酸-三乙胺溶液和洗脱能力强的乙腈对厄贝沙坦进行质量控制和杂质检查[12-13];文献[14-16]报道则使用占较大比例的乙腈来分离厄贝沙坦及相关物质,毒性较大。本研究采用绿色环保、水相约占体积分数90%的微乳液作为流动相,大大减少了有机溶剂的使用,能快速、有效地测定厄贝沙坦及有关物质的质量分数,为厄贝沙坦及其制剂的质量控制提供有效可行的分析方法。
1 仪器与试药
Shimadzu HPLC色谱系统(包括LCsolution液相色谱数据工作站、SPD-20A紫外检测器、Shimadzu LC-20AB泵);AT-330柱温箱(天津奥特赛恩斯仪器有限公司);Agela technologies odyssil C18柱(4.6 mm ×250 mm,5 μm);AKQ-100超声波清洗器(昆山市超声仪器有限公司,频率:40 kHz;功率:100 W);FA1004N电子分析天平、JZ8002分析天平(上海精密科学仪器有限公司);PHS-3C精密酸度计(广州科域新材料科技有限公司)。
厄贝沙坦对照品(美国药典对照品,批号: GOH216);厄贝沙坦有关物质A对照品(美国药典对照品,批号:F11362,质量分数大于99.8%);厄贝沙坦片(本校药物制剂教研室制备,规格:150 mg/片,批号:201307231、201307232、201307233;300 mg/片,批号:201309231、201309232、201309233);厄贝沙坦原料(浙江普洛家园药业公司,批号:1018-1304003V);甲醇、乙腈(色谱纯,美国J.T.Baker公司);聚氧乙烯月桂醚 (Brij35,质量分数99.9%,Amresco公司);十二烷基硫酸钠(SDS,分析纯,天津市福晨化学试剂厂);其他试剂均为分析纯;水为蒸馏水(屈臣氏集团)。
2 方法与结果
2.1溶液的配制
微乳的制备:将SDS、正丁醇、环己烷、水按质量分数3.0%、6.0%、0.8%、92.2%比例依次混合,超声溶解,并调节pH至4.0。
厄贝沙坦对照品储备液的制备:精密称取厄贝沙坦对照品50.01 mg,置于100 mL容量瓶中,加入甲醇溶解并稀释至刻度,制成质量浓度为0.500 1 mg/mL的厄贝沙坦对照品储备液。
厄贝沙坦对照品溶液的制备:精密量取上述厄贝沙坦对照品储备液2 mL,置于10 mL容量瓶中,加入甲醇溶解并稀释至刻度,配制成质量浓度0.100 0 mg/mL的厄贝沙坦对照品溶液。
供试品溶液的制备:取本品 10片(批号: 201309231),约2.7 g,精密称定,研细,精密称取0.018 1 g(相当于10.01 mg厄贝沙坦),置于100 mL容量瓶中,加甲醇配制成厄贝沙坦质量浓度0.100 1 mg/mL的供试品溶液。
有关物质A对照品储备液的制备:取有关物质A 10 mg,精密称定,置500 mL容量瓶中,加甲醇溶解稀释至刻度,得0.020 1 mg/mL的对照品溶液。再精密吸取上述对照品溶液5 mL,置100 mL容量瓶中,制成质量浓度1.005 0 μg/mL的有关物质A对照品储备液。
有关物质A对照品溶液的制备:精密量取有关物质A储备液2 mL,置10 mL容量瓶中,加甲醇稀释至刻度,摇匀,制成质量浓度为0.201 0 μg/mL的有关物质A对照品溶液。
系统适用性溶液:分别取有关物质A和厄贝沙坦对照品各3.5 mg,精密称定,置100 mL容量瓶中,加甲醇稀释至刻度,摇匀,分别配制得有关物质A、厄贝沙坦对照品质量浓度0.035 0mg/mL的系统适用性溶液。
2.2色谱条件与系统适用性试验
色谱柱:Agela technologies odyssil C18柱(4.6 mm ×250 mm,5 μm),流动相:SDS-正丁醇-环己烷-水(质量比3.0∶6.0∶0.8∶92.2),柱温:30℃,流速:1.0 mL/min,进样量:20 μL,检测波长:230 nm。精密吸取“2.1”项下的系统适用性溶液20 μL,注入液相色谱仪,重复进样6次,按上述色谱条件测定并记录色谱图。6次测定结果中厄贝沙坦和有关物质A峰面积的RSD值分别为1.1%和1.2%,均小于2.0%;保留时间的RSD值分别为0.8%和0.7%,小于1.0%;拖尾因子的 RSD值分别为0.8%和0.6%,小于1.0%;有关物质A跟厄贝沙坦峰的分离度大于5.0,均符合质量标准要求,说明本方法的系统适用性良好。
2.3 有关物质的测定
2.3.1专属性试验
2.3.1.1 空白对照试验 取处方配比空白辅料混合粉末适量,精密称定,用甲醇溶解配制得0.1 mg/mL的空白辅料溶液,分别精密吸取空白溶剂、空白辅料溶液、“2.1”项下的供试品、系统适用性溶液各20 μL,注入液相色谱仪,按“2.2”项下条件测定,色谱图见图1。结果显示空白溶剂和空白辅料无干扰。
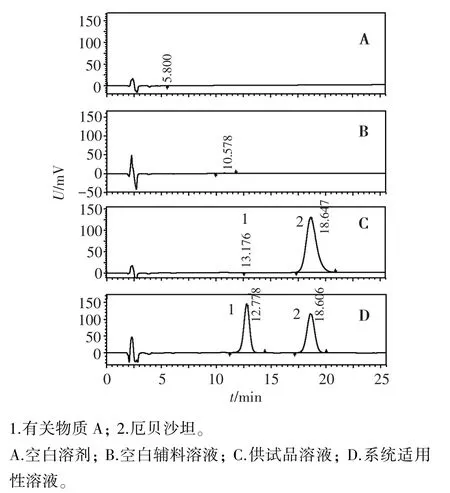
图1 厄贝沙坦专属性试验的HPLC色谱图Figure 1 HPLC chromatograms of irbesartan tablets forspecificity test
2.3.1.2 酸、碱、氧化破坏试验 精密称定厄贝沙坦样品(批号:201307231)5片,共1.335 0 g,平均分成3份,每份0.044 5 g(相当于厄贝沙坦25.00 mg),分别置于25 mL容量瓶中,分别精密加入1 mol/L HCl溶液、1 mol/L NaOH溶液、3%(体积分数)H2O2溶液各5 mL,超声至均匀,避光静置24 h。加甲醇稀释至刻度,分别取1 mL置10 mL容量瓶中,酸、碱破坏溶液分别调pH至中性,3份溶液分别加甲醇稀释至刻度,作为酸、碱、氧化破坏试验溶液。再分别取空白辅料、原料药各25 mg,按照上述步骤操作,分别得到空白辅料、原料药的破坏试验溶液。分别精密吸取20 μL,注入高效液相色谱仪,按“2.2”项下条件测定,空白辅料,原料药,样品的酸、碱、氧化破坏色谱图见图2。结果显示本品在酸条件下没有产生明显的杂质,碱和氧化条件下样品杂质有所增加,其中2个未知杂质(图2B的a和b)为厄贝沙坦在碱破坏条件下的降解产物,碱和氧化破坏均产生了有关物质A(图2中的杂质c),所有杂质峰均能与厄贝沙坦峰完全分离,且溶剂和辅料对有关物质的测定不干扰,说明本方法专属性良好。

图2 酸(A)、碱(B)、氧化(C)破坏试验的HPLC色谱图Figure 2 HPLC chromatograms of irbesartan tablets for damage tests of acid(A),alkali(B)and oxidative(C)
2.3.2线性范围的考察 分别精密量取“2.1”项下质量浓度1.005 0 μg/mL的厄贝沙坦有关物质A储备液0.75、1.0、1.5、2、2.5、3、3.5 mL,分别置于10 mL容量瓶中,加入甲醇稀释至刻度,配制成质量浓度分别为0.075 3、0.100 5、0.150 8、0.201 0、0.251 2、0.301 5、0.351 8 μg/mL的系列对照品溶液。按“2.2”项下色谱条件测定,以厄贝沙坦有关物质A峰面积(A)为纵坐标,溶液质量浓度(ρ)为横坐标进行线性回归,计算得线性方程为A=8.163 4×104ρ+ 204.42,r=0.999 4(n=6)。说明厄贝沙坦有关物质A在0.075 3~0.351 8 μg/mL范围内与峰面积线性关系良好。
2.3.3精密度试验 精密吸取“2.1”项下的厄贝沙坦对照品溶液20 μL注入液相色谱仪,重复进样6次,按“2.2”项下色谱条件测定并记录色谱图。结果厄贝沙坦峰面积的RSD值为1.0%,小于2.0%,表明仪器精密度良好。
2.3.4重复性试验 取同一批厄贝沙坦片(批号: 201307231)适量,共6份,分别按“2.1”项下方法制备供试品溶液,分别精密吸取20 μL进样,按“2.2”项下色谱条件测定。结果6份供试品溶液中厄贝沙坦有关物质A质量分数的RSD值为1.8%,表明本方法重复性良好。
2.3.5稳定性试验 取同一批厄贝沙坦片(批号: 201307231),按“2.1”项下方法制备2份厄贝沙坦供试品溶液,分别贮存在室温与冰箱冷藏室(4℃)中,在0、2、4、6、8、10、16 h分别精密吸取20 μL注入液相色谱仪,按“2.2”项下方法测定。结果室温下有关物质A、最大杂质、总杂质峰面积的RSD值分别为1.51%、4.10%、2.83%,4℃下有关物质A、最大杂质、总杂质峰面积的RSD值分别为5.49%、1.37%、1.78%,均小于10%,表明供试品溶液在室温下放置16 h稳定性良好。
2.3.6回收率试验 取厄贝沙坦片(批号:201307231)10片,精密称定,总质量为2.684 5 g,研细,称取约0.017 9 g(相当于10.00 mg厄贝沙坦),共9份,3份为1组,分别置于100 mL容量瓶中。每组分别加入“2.1”项下1.0050 μg/mL的有关物质A对照品储备液10、20、30 mL,加甲醇定容至刻度,制得有关物质 A质量浓度分别为0.100 5、0.201 0、0.301 5 μg/mL的供试品溶液。分别取上述供试品溶液20 μL进样,依法测定,按外标法以峰面积计算,得3组溶液中有关物质A的回收率分别为101.1%、100.3%、100.1%(n=3),平均回收率为100.5%,RSD=1.1%,表明本方法准确可靠。
2.4厄贝沙坦质量分数的测定
2.4.1线性范围的考察 精密量取“2.1”项下质量浓度0.500 1 mg/mL的厄贝沙坦对照品储备液1.0、1.5、2、2.5、3、3.5 mL,分别置于10 mL容量瓶中,用甲醇稀释至刻度,分别配制成质量浓度为50.01、75.02、100.02、125.02、150.03、175.04 μg/mL的系列厄贝沙坦对照品溶液。按“2.2”项下色谱条件测定,以厄贝沙坦峰面积(A)为纵坐标,溶液质量浓度(ρ)为横坐标进行线性回归,计算得线性方程为A=8.116 8×104ρ+ 2.612 75×104,r=0.999 2(n=6)。结果表明,厄贝沙坦在50.01~175.04 μg/mL质量浓度范围内与峰面积线性关系良好。
2.4.2重复性试验 同“2.3.4”项下操作,结果显示6份供试品溶液中厄贝沙坦质量分数的 RSD为0.6%,表明本方法重复性良好。
2.4.3稳定性试验 同“2.3.5”项下方法操作,室温下和冷冻下厄贝沙坦峰面积的RSD值分别为0.75% 和0.65%,表明供试品溶液在室温放置16 h稳定。
2.4.4检测限与定量限的测定 取0.100 0 mg/mL的厄贝沙坦对照品溶液,逐步稀释,分别注入高效液相色谱仪,依法测定并记录色谱图,色谱图中主峰峰高约为基线噪声3倍(S/N=3)时记为检测限,主峰峰高约为基线噪声10倍时(S/N=10)记为定量限。结果得厄贝沙坦检测限为0.02 μg/mL,定量限为0.1 μg/mL。
2.4.5回收率试验 取厄贝沙坦片(批号:201307231)10片,精密称定,总质量为2.676 1 g,研细,称取约0.008 9 g(相当于4.99 mg厄贝沙坦),共9份,3份为1组。分别置于100 mL容量瓶中,每组分别加入“2.1”项下0.500 1 mg/mL厄贝沙坦对照品储备液6、10、14 mL,加甲醇定容至刻度,制得厄贝沙坦质量浓度分别为79.91、99.91、119.91 μg/mL的供试品溶液。分别精密吸取上述供试品溶液20 μL进样,依法测定,按外标法以峰面积计算,得3组溶液中厄贝沙坦的回收率分别为99.7%、101.0%、100.6%(n=3),平均回收率为100.4%,RSD值为1.0%,表明本方法准确可靠。
2.5样品中厄贝沙坦与有关物质的测定
分别取6 批厄贝沙坦片 (201309231、201309232、201309233、201309231、201309232、201309233),按“2.1”项下供试品溶液的制备方法操作,每批平行操作3次,分别取各供试品溶液20 μL进行测定。结果显示有关物质A、单个杂质、总杂质质量分数分别在0.2%、0.2%、0.5%范围内,2个规格6批样品的有关物质检查均合格。6批样品中厄贝沙坦质量分数均在95.0%~105.0%范围内,样品质量分数符合规定。见表1。
3 讨论
3.1检测波长的选择
制备相同质量浓度的空白辅料、厄贝沙坦、有关物质A溶液和空白溶剂,在200~400 nm进行紫外扫描,厄贝沙坦在205 nm处有较强吸收,在220、230 nm处的吸收也较大;但在205、220 nm处辅料有干扰。而230 nm处厄贝沙坦与有关物质A的吸收均较大,因此选择230 nm为本品的检测波长。
3.2表面活性剂的选择
考察了SDS和Brij35作为表面活性剂对分离的影响,Brij35作为表面活性剂洗脱能力相对较弱,厄贝沙坦和有关物质可达到分离,但分析时间太长。而SDS的洗脱能力较强,厄贝沙坦和有关物质可以分离且分析时间较短,故选择SDS为表面活性剂。
表1 6批厄贝沙坦片中有关物质检查和厄贝沙坦质量分数测定结果Table 1 Determination results of irbesartan and the related substances in irbesartan tablets of two specifications of 6 batches of samples(s) w/%

表1 6批厄贝沙坦片中有关物质检查和厄贝沙坦质量分数测定结果Table 1 Determination results of irbesartan and the related substances in irbesartan tablets of two specifications of 6 batches of samples(s) w/%
规格 批号 有关物质A 最大杂质 总杂质 厄贝沙坦 平均标示量150 mg/片 201307231 0.12±0.9 0.14±1.1 0.26±1.1 98.7±1.1 99.4±0.6 201307232 0.07±0.7 0.14±1.0 0.21±0.9 99.6±0.6 201307233 0.15±0.7 0.15±0.8 0.30±1.0 100.0±0.2 300 mg/片 201309231 0.09±1.1 0.14±1.0 0.26±0.8 99.7±0.6 99.8±0.4 201309232 0.12±0.6 0.13±0.8 0.25±1.0 99.0±0.2 201309233 0.08±0.8 0.10±1.1 0.18±1.2 100.7±0.4
3.3油相的选择
考察了正辛醇、环己烷、正己烷、正庚烷作为油相对分离的影响,结果显示对厄贝沙坦和有关物质的洗脱能力由小到大依次排列为正辛醇<环己烷<正己烷<正庚烷。正庚烷和正己烷的洗脱能力太强,不适合分离,而正辛醇的洗脱能力太弱,分析时间太长,环己烷的洗脱能力适中,适合厄贝沙坦和有关物质的分离;故选择环己烷作为油相。
3.4流动相pH的选择
采用SDS-正丁醇-环己烷-水(质量比3.0∶6.0∶0.8∶92.2)为微乳流动相,分别考察pH值为3、4、5、6时对分离选择性的影响,结果表明:pH为3时,保留因子较大,分离时间过长;pH为5时,保留降低,不利于分离且柱效较低;pH为4时,柱效较高且分离时间较短,故调节流动相pH值为4。
本研究采用微乳液相色谱法分离厄贝沙坦和有关物质,通过对检测波长、表面活性剂、油相、流动相pH的选择,确定了最佳微乳流动相体系,并进行了方法学考察,结果表明本方法快速、准确、重复性好,可用于厄贝沙坦和有关物质的分离检测。
[1]黄震华.血管紧张肽Ⅱ受体拮抗药厄贝沙坦在高血压治疗中的应用[J].中国新药与临床杂志,2007,26(5): 384-387.
[2]蔡思宇,张雪华,胡晓晟,等.厄贝沙坦治疗轻、中度高血压病[J].中国新药与临床杂志,2002,21(5):277-280.
[3]孙慧君.厄贝沙坦治疗2型糖尿病肾病的临床疗效观察[J].临床医学工程,2010,17(6):28-29.
[4]ZANG Ruirui,CHEN Xiaohui,LI Qing,et al.Liquid chromatography coupled with mass spectrometry method for thesimultaneousquantificationofirbesartanand hydrochlorothiazide in human plasma[J].Chin Pharma Sci,2011,20(4):360-367.
[5]QIU Xiangjun,WANG Zhe,WANG Bing,et al.Simultaneous determination ofirbesartanandhydrochlorothiazidein human plasma by ultra high performance liquid chromatography tandem mass spectrometry and its applicationto a bioequivalence study[J].J Chromatogr B,2014,957(1): 110-115.
[6]魏献波,王美艳,路婷婷.高效液相色谱法测定厄贝沙坦的含量[J].中国生化药物杂志,2015,35(10):141-143.
[7]熊思敏,吕竹芬,谢清春,等.高效液相色谱法测定厄贝沙坦缓释片的含量[J].中国药师,2008,11(2):205-207.
[8]益磊,施志顺,周志美,等.HPLC法测定厄贝沙坦胶囊中厄贝沙坦的含量[J].中国医药科学,2012,2(2):118-120.
[9]李家明,吴强,王玲玲.HPLC法测定厄贝沙坦片剂的含量[J].药物分析杂志,2001,21(4):249-250,308.
[10]SHAKYA A K,AL-HIARI Y M,ALHAMAMI O M.Liquid chromatographic determination of irbesartan in human plasma[J].J Chromatogr B,2007,848(2):245-250.
[11]SHRIKANT H P,MINAKSHI V J.Novel and validated titrimetricmethodfordeterminationofselected angiotensin-Ⅱ-receptorantagonistsinpharmaceutical preparations and its comparison with UV spectrophotometric determination[J].J Pharm Anal,2012,2(6): 470-477.
[12]国家药典委员会.中华人民共和国药典:2015年版二部[M].北京:中国医药科技出版社,2015:72-73.
[13]The United States Pharmacopieial Convention.USP[M]. American:UnitedStatesPharmacopoeiaAssociation,2012:5555-5556.
[14]孙启泉,刘燕,施介华,等.RP-HPLC法测定厄贝沙坦的含量及有关物质[J].浙江化工,2006,37(6):20,28-29.
[15]梁艳芬,许丽媛,黎亮星,等.HPLC测定厄贝沙坦片有关物质[J].食品与药品,2015,17(4):235-240.
[16]江东波,马晓鹂,蔡伟明,等.RP-HPLC法测定厄贝沙坦缓释胶囊中主药及有关物质的含量[J].中国药房,2009,20(4):286-287.
(责任编辑:刘晓涵)
Determination of irbesartan tablets and related substances by microemulsion liquid chromatography
ZHENG Aini,WANG Liuyun,YU Nuojun,LI Ning
(School of Pharmacy,Guangdong Pharmaceutical University,Guangzhou 510006,China)
Objective A microemulsion liquid chromatography method was developed for the determination of irbesartan and related substances in tablets.Methods Chromatographic separation was performed on a Agela technologies odyssil C18(4.6 mm×250 mm,5 μm)at the column temperature of 30℃.The mobile phase was SDS-butylalcohol-cyclohexane-water(3.0∶6.0∶0.8∶92.2)at a flow-rate of 1.0 mL/min and the injection volume was 20 μL.The detection wavelength was 230 nm.Results Related substances were completely separated from the major constituents.In the range of 50.01 to 175.04 μg/mL(r=0.999 2,n= 6),it has a good linear relationship for irbesartan with the peak area.The average recovery of irbesartan was 100.4%(RSD=1.0%,n=9)and it′s detection limit was 0.02 μg/mL.While the related compound A was linear over the range of 0.075 3-0.351 8 μg/mL,with a detection limit of 0.025 μg/mL and it′s average recovery was 100.5% (RSD=1.1%,n=9).The contents of 2 specifications were 99.4%and 99.8%,respectively.Conclusion A rapid,accurate and reproducible determination method of irbesartan and related substances with microemulsion liquid chromatography was established.It was green and environmental,and could reduce the use of organic solvents.
irbesartan tablets;MELC;irbesartan;related substance
R927.2
A
1006-8783(2016)04-0444-05
10.16809/j.cnki.1006-8783.2016041305
2016-04-13
国家自然科学基金(81173525)
郑艾妮(1992—),女,2015级硕士研究生,主要从事药物食品分析与质量控制研究,Email:13694237621@163.com;通信作者:李宁(1959—),女,教授,主要从事天然药物及化学药物质量标准研究,电话:020-39352136,Email: 13724117338@163.com。
网络出版时间:2016-07-06 10:39 网络出版地址:http://www.cnki.net/kcms/detail/44.1413.R.20160706.1039.002.html
