基于高通量测序对宜宾芽菜中细菌群落结构分析
2016-09-10王小龙叶碧霞杨小龙
左 勇,王小龙,叶碧霞,江 鹏,傅 彬,杨小龙
(四川理工学院生物工程学院,四川自贡 643000)
基于高通量测序对宜宾芽菜中细菌群落结构分析
左勇,王小龙,叶碧霞,江鹏,傅彬,杨小龙
(四川理工学院生物工程学院,四川自贡 643000)
目的:研究宜宾芽菜发酵过程中细菌微生物多样性。方法:采用Illumina高通量测序技术对芽菜样本中所有细菌的16S V1-V3区进行测序,应用QIIME软件和Mothur软件分析和统计样品序列数目、OUT数量,并进行聚类分析。结果:宜宾芽菜发酵过程中6个阶段的样本共获得204997条有效序列,优化后有效序列数为41703条,共9202个OTU分类;主要细菌类群为Firmicutes(厚壁菌门)下的Bacillus(芽孢杆菌属),Bacteroidetes(拟杆菌门)下的Flavobacteriaceae(黄杆菌属)和Sphingobacterium(鞘脂杆菌属),Proteobacteria(变形菌门)下的Sphingomonas(鞘脂单孢菌属)、Acidovorax(嗜酸菌属)、Chromohalobacter(色盐杆菌属)和Pseudomonas(假单胞菌属)以及Actinobacteria(放线菌门)下的Arthrobacter(节杆菌属)。结论:宜宾芽菜发酵过程中细菌菌群处于动态变化中,随着发酵的进行,部分细菌逐渐减少或消失,优势细菌趋于稳定。
宜宾芽菜,细菌群落,高通量测序,Miseq
宜宾芽菜是由芥菜的嫩茎划成条状,在大量微生物的作用下,经过自然发酵腌制而成,是我国加工食品中非常著名的产品,有川菜“四大名菜”之称,成为四川家喻户晓的传统酱腌菜[1]。芽菜发酵过程中主要功能菌为细菌,据研究表明,在芽菜发酵过程中有大量的乳酸菌类、嗜盐、嗜酸类细菌[2-3]。它们之间互营共生,生成芽菜主要的风味物质和营养物质,因此研究芽菜发酵过程中细菌的群落结构将对提高宜宾芽菜品质有重要作用。
传统的研究芽菜中微生物的方法是菌种分离,近几年开始运用分子生物学方法对对芽菜中微生物进行研究,如PCR-DGGE技术[4]。传统的菌种分离只能对芽菜中极少数能分离的菌种进行研究,PCR-DGGE技术对芽菜的研究无法准确的定量,也不能对样品中微生物群落进行较为完备的测序研究,同时工作量大、灵敏度不高。相比较这些方法,高通量测序(HTS)技术(又称“下一代”测序(NGS)技术)对微生物群落结构的研究有其明显的先进性和优势,准确定量、读长较长、实时检测等。目前,高通量测序技术已经开始运用于各种分子生物学领域[5],在人体微生物、水中生物、土壤、热泉微生物[6-9]等多个领域都有应用。
本研究首次运用高通量Illumina MiSeq 2 x 300 bp测序平台对宜宾芽菜发酵过程中的细菌进行研究,建立一套高通量测序技术分析宜宾芽菜细菌的方法,同时利用QIIME和Mothur软件相结合,运用于数据分析,能够更加准确、完整地解析宜宾芽菜中细菌的群落结构,为进一步利用和开发宜宾芽菜中微生物资源提供研究基础。
1 材料与方法
1.1材料与仪器
芽菜样本采自宜宾双谊富康芽菜厂,分别为同一批次发酵15 d(编号1)、30 d(编号2)、60 d(编号3)、90 d(编号4)、120 d(编号5)、150 d(编号6),迅速置于冰盒运回,-20 ℃保藏;TaqDNA polymerase、dNTPs、DL2000TM DNA Marker宝生物工程(大连)有限公司;蛋白酶KMerck;溶菌酶 Sigma;琼脂糖、丙烯酰胺、N,N′-亚甲基双丙烯酰胺、去离子甲酰胺 Solarbio;引物由上海英潍捷基生物技术公司合成。
通用型恒压恒流电泳仪北京君意东方电泳设备有限公司;My Cycler型PCR仪美国Bio-rad公司;Bio-Best 200E型凝胶成像分析系统美国SIM公司;高速冷冻离心机X1R美国Thermo公司;台式冷冻离心机Eppendorf;MX-S型可调式混匀仪美国赛洛捷克;SW-CG-1F型超净工作台苏州苏洁净化设备有限公司;微量荧光核酸定量仪Qubit 2.0;Miseq 测序仪Illumina公司。
1.2总DNA的提取及PCR扩增
采用液氮研磨+溶菌酶+SDS高盐抽提法[10]提取芽菜中微生物总宏基因组,然用核酸蛋白仪检测DNA的浓度和纯度,置于-20 ℃备用。
通用引物选择序列较长的V1-V3序列F8-R533[11],因其扩增效果好,通用性较高,可以扩增出650 bp左右的片段。PCR反应体系为25 μL,体系包括5 μL 5×Buffer,0.6 μL 10 mmol/L dNTP,0.5 U phusion超保真DNA 聚合酶,DNA 模板用量为2 μL,533R内侧引物(5 pmol/L)2 μL,8F内侧引物10 μmol/L母液稀释48倍取1 μL,8F外侧引物10 μmol/L母液稀释1.6倍取1 μL,533R外侧引物10 μmol/L母液稀释1.6倍取1 μL,补水至25 μL。PCR 反应程序为:98 ℃ 30 s;98 ℃10 s,50 ℃ 90 s,72 ℃ 30 s,8个循环;72 ℃ 5 min;98 ℃ 10 s,50 ℃ 90 s,72 ℃ 30 s,72 ℃ 30 s,30个循环;72 ℃ 5 min;10 ℃ 10 min。PCR结束后取3 μL PCR产物于1.2%的琼脂糖凝胶电泳检测。
一个星期后,斯通只身潜水进入奥古斯丁聚水坑,去重新完成伊恩和肯尼中断的探险任务。中间集结营地有一支后援队作好了准备,他就游回到那充满空气的石室。为了纪念伊恩·罗兰,探察队已将这石室命名为“罗兰气钟”。
1.3PCR产物纯化及定量
采用QIAquick Gel Extraction Kit胶回收试剂盒对PCR扩增产物进行切胶回收,取3 μL回收产物进行电泳检测。
采用Quant-iT PicoGreen 定量试剂盒[12]对胶回收之后的PCR产物进行定量。
1.4高通量测序
将定好量的DNA文库样本送至微基生物科技(上海)有限公司进行高通量测序。
1.5QIIME软件和Mothur软件进行生物信息学分析
2 结果与分析
2.1样本文库的获得及定量
提取6个芽菜样本总DNA,对细菌16S rRNA V1-V3区基因扩增,PCR扩增结果进行胶回收,获得样本文库(见图1、图2)。由图1可知,基因组条带可见,部分DNA样本浓度低,条带较弱,均可正常进行后续实验;由图2可见,电泳检测条带单一,片段长度与预期片段大小一致,约为650 bp,且浓度适中,可用于后续实验。

图1 芽菜样本总DNA的琼脂糖凝胶电泳Fig.1 Sprouts samples of total DNA agarose gel electrophoresis注:1~6分别代表芽菜发酵时间15、30、60、90、120、150 d;M:DL2000TM DNA Marker,图2同。
图2 芽菜样本细菌PCR扩增结果(A)和胶回收结果(B)Fig.2 Sprouts sample bacteria PCR amplification (A)and gel recovery results(B)
采用微量荧光核酸定量仪Qubit 2.0对胶回收产物进行定量。从图3可以看出,标准曲线符合定量标准,以此计算的定量结果较为准确,表1为由此标准曲线计算出的各样品PCR产物定量的浓度,定量浓度满足高通量测序要求。

图3 胶回收产物定量标准曲线Fig.3 Gel product recycling quantitative standard curve
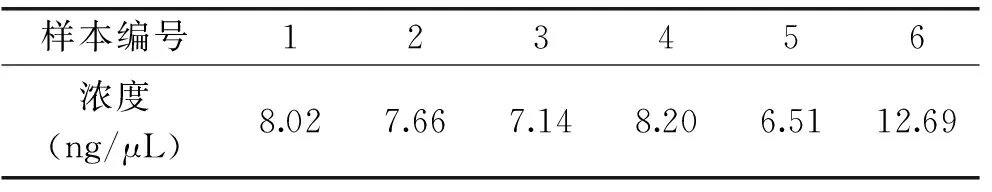
样本编号123456浓度(ng/μL)8.027.667.148.206.5112.69
2.2细菌群落的OUT分类
将6个芽菜样本进行高通量测序,根据barcode标签序列筛选出有效序列,并对处理后的有效序列进行数据及长度分布统计,细菌共获得41703条有效序列。根据97%水平的OTU丰度,使用rarefaction curve对样本多样性进行评估,得到样本稀释曲线。据图4(A)中所示,随着测序深度的增加,各样本的曲线斜率逐渐减小,最终趋于平缓。说明测序深度能够较准确地反映芽菜中细菌的丰富度和多样性。利用各样本的测序量在不同测序深度时的细菌多样性指数构建Shannon-Wiener曲线,以此反映各样本在不同测序数量时的微生物多样性[14]。从图4(B)中可以看出,各样本曲线趋向平坦,说明测序数据量足够大,可以反映样本中绝大多数的微生物物种信息。
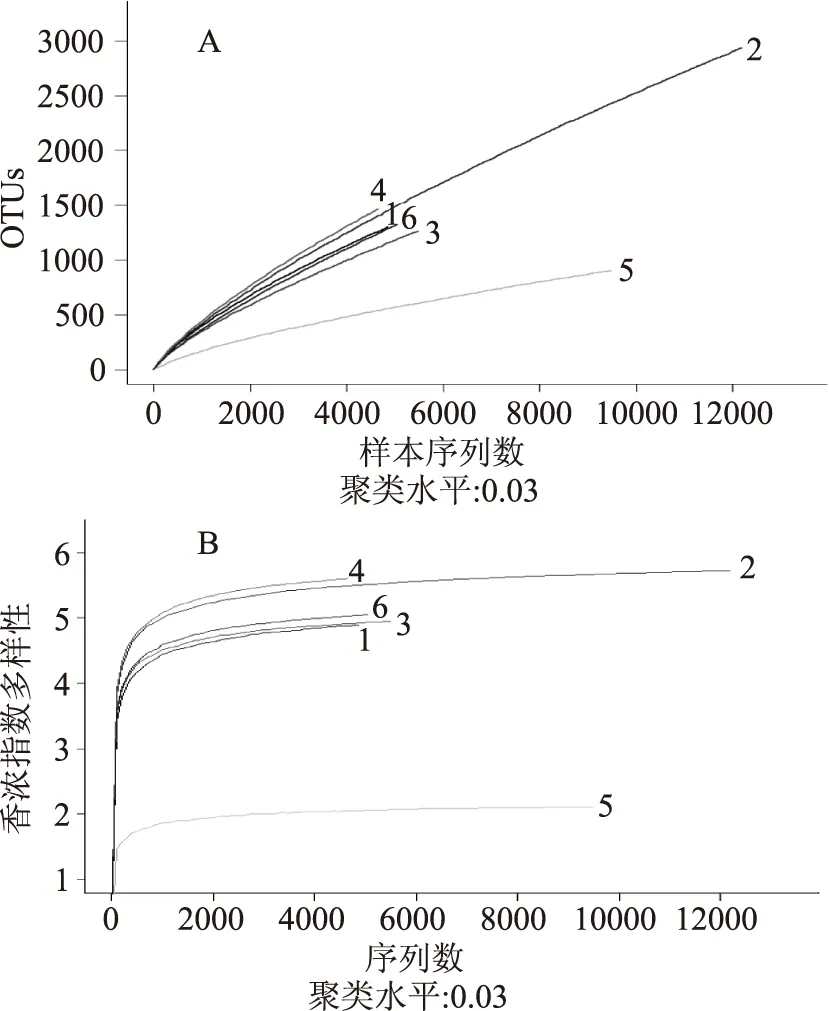
图4 测序深度曲线Fig.4 Sequencing depth curve注:A:稀释性曲线,B:Shannon-Wiener曲线。
2.3各样本在属水平上的比较
6个样本的有效细菌基因序列录入BLAST结合MEGAN软件,在属分类水平上分析,根据各样本中的细菌组成绘制柱状图(图5),比较各样本在属分类水平上群落结构组成的差异。结果显示,发酵120 d和150 d的优势菌与其他4个时期有较大差别,其中发酵120 d时有超过90%的序列为Chromohalobacter,发酵150 d时占优势的细菌为Lactobacillus(44%)、Pseudomonas(15%)Pseudomonas(14%)。其他发酵时间段的优势细菌分别为:发酵15 d时占优势的细菌为Kushneria、Unclassified、Buttiauxella、Pseudomonas、Sphingomonas;发酵30 d的优势细菌为Kushneria、Unclassified、Pseudomonas、Pectobacterium、Methylobacterium、Arthrobacte、Sphingomonas;发酵60 d的优势细菌为Unclassified、Serratia、Sphingomonas、Rhizobium;发酵90 d的优势细菌为Unclassified、Buttiauxella、Exiguobacterium、Leuconostoc、Staphylococcus、Arthrobacter、Enterobacter。
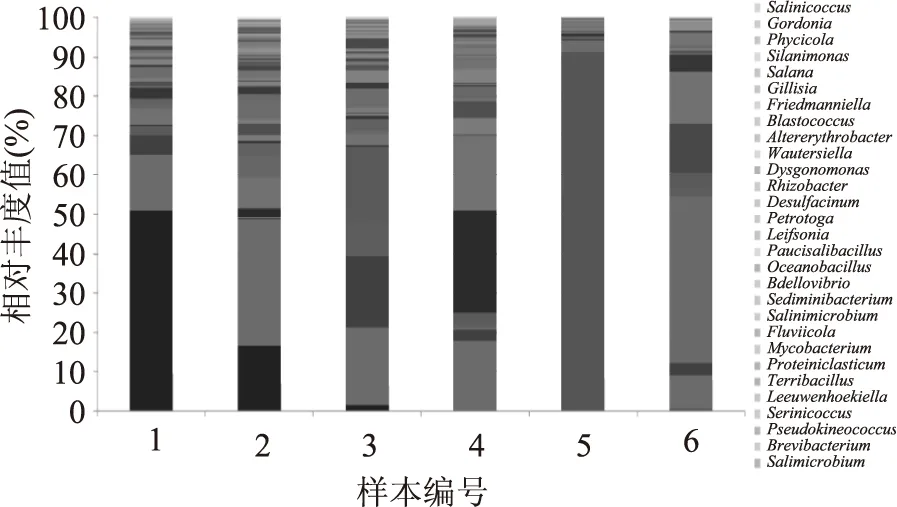
图5 各样本在属水平上群落结构组成的差异Fig.5 All the samples in the genus level of community structure
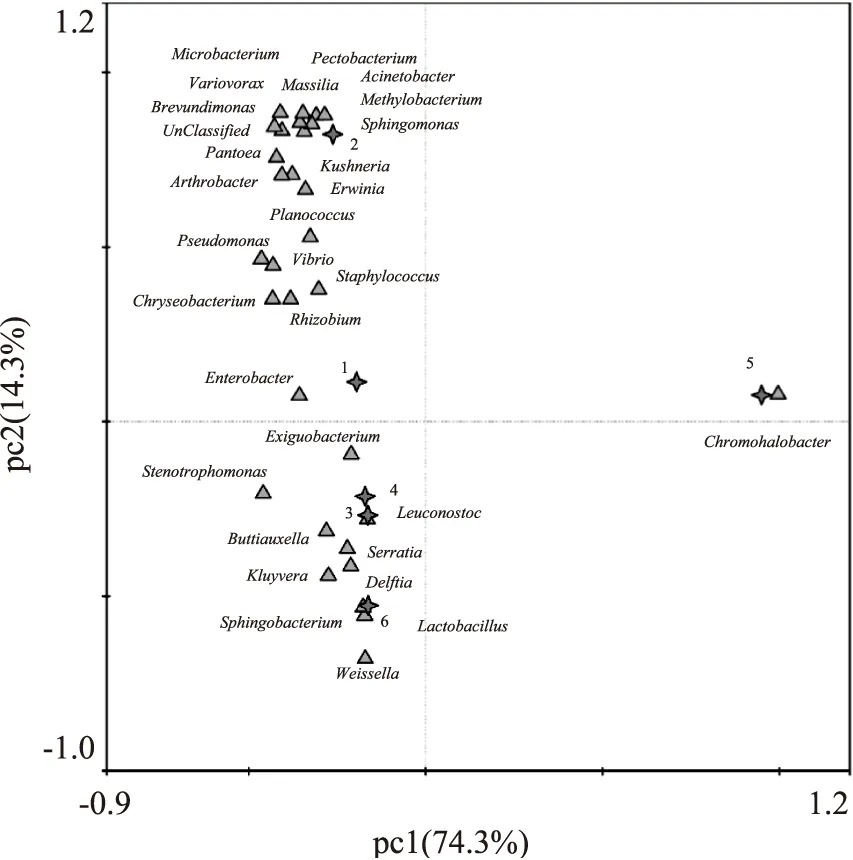
图6 基于属水平的PCA图Fig.6 Based on the genus level of PCA figure
由各样本OUT组成的差异,通过方差分解,作出PCA图(图6)。PC1和PC2的贡献率分别为74.3%和14.3%,累计贡献率高达88.6%,是变异的主要来源,可以解释变量的绝大部分信息。由图6可知,芽菜发酵120 d时细菌的多样性与其他发酵时期有较大差别,主要是因为Chromohalobacter所占的比例较大,发酵前期与发酵后期细菌的组成也有较大差异性,由此可知芽菜发酵过程中优势细菌的组成是一个动态变化的过程,随着发酵的进行,部分细菌逐渐减少或消失,优势细菌逐渐趋于稳定,含量有所差异。
2.4基于UniFrac的聚类的比较分析
分析了宜宾芽菜6个不同发酵时期样本中细菌菌群的总体结构,对比分析6份芽菜样本间菌群多样性的相似度,可以采用2种图(树图和热点图)来表示。其中比对得到的树图(图7),图中显示所有枝条的结点表示相邻两个细菌多样性有显著性差异(p<0.01),特别是5号样本与其他样本有明显菌群的差异,除5号样本其他样本都依次按照发酵时间相邻。
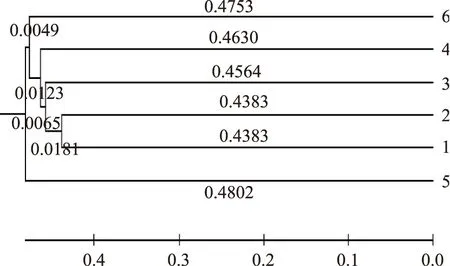
图7 全样本相似性分析树图Fig.7 All sample tree graph similarity analysis
将指定种属水平上的分类信息(本次绘图是根据genus水平上OUTsize位于前50的信息进行绘制的)分别按照样品和分类进行聚类后作出Heatmap图(图8),通过颜色的梯度及相似程度来反映数据的相似性和差异性,能够反映出所有样本在各分类水平上表现的相似性或者差异性。由图8可知1、2、3号样本聚为一类,细菌种类的组成基本相同,但某些细菌的含量在逐渐减少,甚至消失;4、5、6号样本聚为一类,细菌种类趋于稳定,含量有所差异,尤其5号样本色盐杆菌属含量与其他样本有明显差异。说明在宜宾芽菜发酵过程中,发酵前期细菌的种类变化不大,在数量上有部分变化;发酵后期细菌的种类已经比较稳定,在各菌群含量上有所差异。
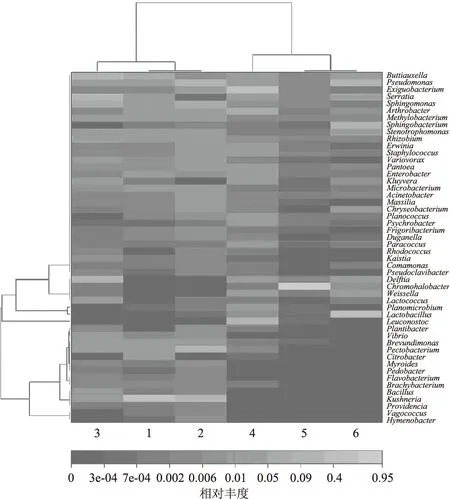
图8 全样本多样性热图Fig.8 All samples diversity heatmap
3 结论
通过Miseq测序方法全面的分析了宜宾芽菜发酵过程中细菌的多样性,结果显示6个阶段的样本共获得204997条有效序列,优化后有效序列数为41703条,共9202个OTU分类。通过芽菜样本的细菌主成分分析结果,得出发酵各时期的优势细菌,通过热图反映了各样本之间的相似性与差异性。Miseq测序16S rDNA V1-V3序列反映了更多的多样性种类,比以往任何芽菜细菌多样性的研究得到了更多的分类种类和未知属种的细菌,证实了宜宾芽菜在发酵过程中含有丰富的细菌资源。
对芽菜发酵微生物群落结构的研究与传统的培养方法及16S rDNA为基础的分子生物学方法不同,利用高通量测序的方法研究宜宾芽菜发酵过程中细菌群落结构,全面客观地分析了芽菜中的微生物多样性,发现了一些在芽菜中以前未报道的细菌类,如Sphingobacterium(鞘脂杆菌属)、Chromohalobacter(色盐杆菌属)、未培养细菌等,同时本研究分析了各类优势菌的含量,根据数据客观地反映宜宾芽菜发酵过程中各时期的细菌群落结构。本实验中细菌引物设计在650 bp左右,测序数据对不同发酵时期芽菜中微生物的区分度将扩大,能够更好的了解芽菜中微生物的类群,同时建立较为完善的芽菜微生物数据库,为芽菜生产和提高芽菜质量提供了较为准确的参考数据。
[1]徐坤,雷激,李博,等.芽菜腌制过程中理化指标的动态研究[J].中国酿造,2010(1):29-32.
[2]尹曦,蒲彪,陈安均,等.四川宜宾芽菜中乳酸菌分离筛选及菌相分析[J].食品科学,2013(3):163-168.
[3]张先琴,张小平,敖晓琳,等.PCR-DGGE分析四川地区家庭制作泡菜中微生物多样性[J].食品科学,2013(12):129-134.
[4]张先琴,张小平,敖晓琳,等.PCR-DGGE分析四川地区家庭制作泡菜中微生物多样性[J].食品科学,2013(12):129-134.
[5]Maccallum.ALLPATHS 2:small genomes assembled accurately and with high continuity from short paired reads[J]. Genome Biol, 2009, 10(10):R103.
[6]Yang F, Zeng X,Ning K.Saliva microbiomes distinguish caries-active from healthy human populations[J]. The ISME Journal,2011,106(40):172-175.
[7]李晓然.基于核糖体RNA高通量测序分析微生物群落结构[D].上海:复旦大学,2011.
[8]Zhang L, Offre PR, He J,et al. Autotrophic ammonia oxidation by soil thaumarchaea[J]. Proceedings of the National Academy of Sciences,2011,107(40):17240-17245.
[9]Lehtovirta-Morley LE, Stoecker K,Vilcinskas A, et al.Cultivation of an obligate acidophilic ammonia oxidizer from a nitrifying acid soil[J]. Proceedings of the National Academy of Sciences,2011,108(38):15892-15897.
[10]倪峥飞,许伟,窦文芳,等.镇江香醋固态发酵醋醅中微生物总DNA提取方法比较[J].微生物学报,2010(1):119-125.
[11]Sizhong Yang, Xi Wen.Pyrosequencing investigation into the bacterial community in permafrostsoils along the China-Russia crude oil pipeline(CRCOP)[J]. PLoS ONE, 2012, 7(12):e52730.
[12]Acosta-Martmez V,Dowd SE,Sun Y, et al.Pyrosequencing analysis for characterization of soil bacterial populations as affected by an integrated livestock-cotton production system[J]. Applied Soil Ecology,2010,45(1):13-25.
[13]Chun-Lan Xu, Rui Sun,Xiang-Jin Qiao, et al. Protective effect of glutamine on intestinal injury and bacterial community in rats exposed to hypobaric hypoxia environment[J]. World Journal of Gastroenterology,2014(16):4662-4674.
[14]余琼,李东尧,毛丙永,等.麻辣提取物对肠道微生物的影响[EB/OL]. 中国科技论文在线.
Based on high-throughput sequencing analysis of bacterial community structure in Yibin sprouts
ZUO Yong,WANG Xiao-long,YE Bi-xia,JIANG Peng,FU Bin,YANG Xiao-long
(Dept. of Bioengineering,Sichuan University of Science and Engineering,Zigong 643000,China)
Objective:The aim of this study was to analyze the yibin sprouts bacteria microbial diversity in the process of fermentation.Methods:Illumina high-throughput sequencing techno-logy were used to sequence 16S V1-V3 hypervariable reqion of all 6 yibin sprouts samples. QIIME and Mothur was used to analyze and calculate the numbers of sequences and operational taxonomic unitl(OTUs)for each sample,and followed by cluster analysis.Results:Yibin sprouts 6 stages in the process of fermentation samples received 204997 valid sequence,the optimized effectively sequence number was 41703,a total of 9202 OTUs.The main groups for bacteria of Yibin sprouts wasBacillusunder theFirmicutes,FlavobacteriaceaeandSphingobacteriumunder theBacteroidetes,Sphingomonas,Acidovorax,ChromohalobacterandPseudomonasunder theProteobacteria,andArthrobacterunder theActinobacteria.Conclusion:Yibin sprouts bacterial flora in the fermentation process of dynamic change,with the fermentation proceeds,some bacteria gradually decreased or disappeared,dominant bacteria stabilized.
Yibin sprouts;bacterial community;high-throughput sequencing;Miseq
2015-08-03
左勇(1972-),男,硕士,教授,主要从事发酵工程方面的研究,E-mail:sgzuoyong@tom.com。
固态发酵资源利用四川省重点实验室开放基金项目(2015gtc002);2014四川省科技厅农业科技成果转化项目;2013大学生创新创业训练计划项目(201310622001);四川理工学院培育项目(2013PY09)。
TS264.2
A
1002-0306(2016)10-0242-05
10.13386/j.issn1002-0306.2016.10.040
