镁原子与掺杂石墨烯吸附体系的微观结构分析
2016-09-10山西中北大学机电工程学院向丰华
山西中北大学机电工程学院 向丰华
镁原子与掺杂石墨烯吸附体系的微观结构分析
山西中北大学机电工程学院 向丰华
运用密度泛函理论对镁原子与掺杂石墨烯之间的吸附反应进行了第一性原理计算,研究了掺杂原子对石墨烯以及镁原子吸附对掺杂石墨烯的微观结构影响。本文为镁原子与石墨烯的实验研究提供了重要的理论基础,具有重要的实践指导意义。
密度泛函;镁原子;石墨烯;微观结构
引言
镁基复合材料拥有优异的力学和物理性能,但其弹性模量普遍偏低从而极大地限制了其在结构材料领域的广泛应用。石墨烯具有高弹性模量(约1TPa)与断裂强度(约125GPa)[1],将石墨烯作为镁基复合材料的增强相,能有效地提高镁金属及镁合金的强度、弹性模量等性能。因此研究镁与石墨烯的界面反应是该领域研究的重要课题。由于本征石墨烯是带隙为零的半导体[2]且本征石墨烯这种带隙为零的能带结构容易受到各种外在因素的影响,例如空位缺陷、表面吸附、晶格畸变以及掺杂等。这些因素的影响会导致本征石墨烯的带隙打开,同时带来各种独特的性质,从而会对镁与石墨烯的相互作用造成影响。因此,本文主要从硼(B)、氮(N)和硅(Si)三种非金属元素掺杂石墨烯的角度出发,对镁原子与掺杂石墨烯吸附体系的结构性质进行研究。
1 吸附模型与计算参数
1.1 吸附模型的建立
首先,建立一个4×4周期性石墨烯超晶胞,模型中碳碳键长为1.425,超晶胞晶格常数a=b=9.870。掺杂石墨烯通过此4×4周期性石墨烯超晶胞(包括32个碳原子)中置换一个碳原子的方式进行构造的。然后,再通过在掺杂石墨烯表面上加入一个镁原子的方式来构造镁原子与掺杂石墨烯的吸附模型。石墨烯及掺杂石墨烯超晶胞在垂直于石墨烯表面的方向上有30 厚度的真空层,以确保吸附的镁原子只与其中一层石墨烯反应。并用UBER法[3]对镁原子与掺杂石墨烯吸附体系中的吸附初始高度进行了测试。
1.2 计算参数的选定
本文运用Dmol3程序包来进行第一性原理计算。计算过程中选定广义梯度近似(GGA)和PBE密度泛函作为处理计算电子相互作用的交换相关泛函,考虑到研究体系的特性本文计算过程还加入了自旋极化和偶极矫正,并在计算过程中采用高斯展开,其展开宽度设为0.05eV;超晶胞模型中的所有的原子都要进行充分弛豫,直到每个原子所受到的力都小于0.01eV/;碳原子轨道半径=6.0;k-points取值=8×8×1,并且在整个弛豫优化过程中超晶胞的晶格常数保持不变。
1.3 结果分析
1.3.1 掺杂石墨烯结构分析
图1.1为掺杂石墨烯的优化结构,从图中可以看出石墨烯中的碳碳键结构受到了一定程度的改变,包括其键长和键角均发生了一定的变化,尤其是掺杂原子附近的碳碳键。表1.1为掺杂原子与其第一近位碳原子的结合键键长(吸附前)。从中吸附前的键长数据中可以看出相比于本征石墨烯中C-C键长(1.425),掺杂石墨烯中:Si-C键最长(1.654),其次为B-C键(1.484),而N-C键的键长最小(1.415)。
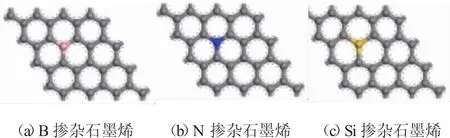
图1.1掺杂石墨烯模型优化结构图
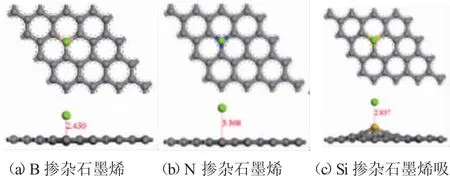
图1.2镁原子与掺杂石墨烯吸附模型优化结构三维视图
表1.1在吸附镁原子前后掺杂原子 。与其第一近邻碳原子之间的键长(单位:)
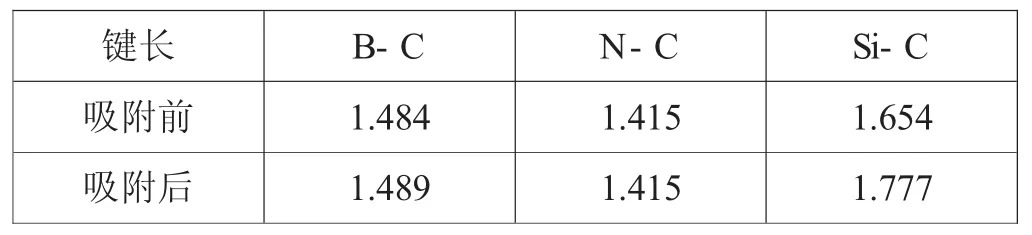
表1.1在吸附镁原子前后掺杂原子 。与其第一近邻碳原子之间的键长(单位:)
键长 B-C N-C Si-C吸附前 1.484 1.415 1.654吸附后 1.489 1.415 1.777
表1.2优化后镁原子与掺杂原子以及其第一近邻碳原子之间的距离(单位:)
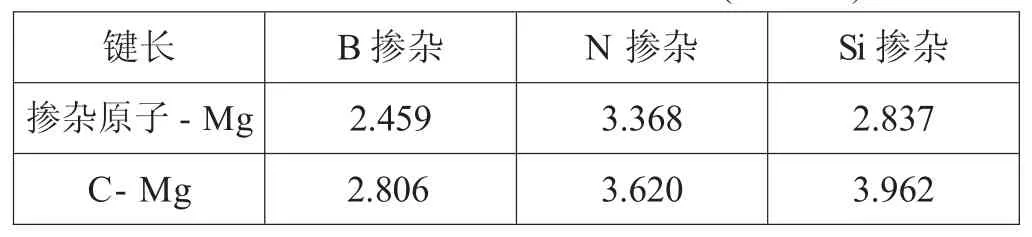
表1.2优化后镁原子与掺杂原子以及其第一近邻碳原子之间的距离(单位:)
键长 B掺杂 N掺杂 Si掺杂掺杂原子-Mg 2.459 3.368 2.837 C-Mg 2.806 3.620 3.962
1.3.2 镁原子在掺杂石墨烯表面吸附模型结构分析
图1.2为镁原子与掺杂石墨烯吸附模型在经过充分弛豫优化后的结构图。从中可以看出在吸附镁原子之后,掺杂石墨烯结构发生了一定的改变。例如图1.2(c),在吸附镁原子后Si掺杂石墨烯中的掺杂原子严重脱离了之前的平衡位置与所在平面。并且,在掺杂原子周围的结合键键长由开始的1.654和1.631,分别变成了1.777和 1.751(表1.1中吸附后)。但在B和N原子掺杂的石墨烯中相应的结合键键长却只发生非常微小的改变。同时,为了进一步分析镁原子与掺杂石墨烯平面的相互作用,本文还计算了镁原子与掺杂原子的吸附距离,即经过充分弛豫优化后的距离。表1.2中可以看出B、N和Si原子掺杂石墨烯与镁原子的吸附模型中,相应的掺杂原子与镁原子之间的距离分别为2.459,3.368和2.837。
2 总结
采用第一性原理计算方法对镁原子与B、N和Si三种原子掺杂石墨烯吸附体系结构特性进行了研究。计算结果表明,镁原子的吸附对B、N原子掺杂石墨烯的晶胞结构影响变化较小而对于S掺杂石墨烯的晶胞结构影响结构较大。掺杂原子对石墨烯与镁原子的吸附强度的增强大小顺序为:B>Si>N。
[1]赵力涛,王文军.石墨烯物理性质的研究进展[J].科技信息,2012(6):165-165.
[2]Geim A K.Graphene:status and prospects.[J].Science,2009,324(5934):1530-1534.
[3]Rose J H,Ferrante J,Smith J R.Universal Binding Energy Curves for Metals and Bimetallic Interfaces[J].Physical Review Letters,1981,47(9):675-678.
