瑞舒伐他汀对急性心肌梗死大鼠心肌细胞凋亡的抑制作用及其机制
2016-09-05杨蕾刘海龙张斌汤建民
杨蕾,刘海龙,张斌,汤建民
(郑州大学第二附属医院,郑州450000)
瑞舒伐他汀对急性心肌梗死大鼠心肌细胞凋亡的抑制作用及其机制
杨蕾,刘海龙,张斌,汤建民
(郑州大学第二附属医院,郑州450000)
目的探讨瑞舒伐他汀对急性心肌梗死(AMI)大鼠心肌细胞凋亡的抑制作用及其机制。方法 从60只SD雄性大鼠中随机抽取10只作为对照组,只分离不结扎前降支,余下的50只分别结扎其前降支制作AMI模型。24 h后存活的41只随机分为3组,分别为AMI组13只、Statin1组14只、Statin2组14只。术后第1天起,Statin1、2组分别给予瑞舒伐他汀1.5、3.0 mg/(kg·d)灌胃,对照组及AMI组分别以等量的生理盐水灌胃。4周后TUNEL法染色检测心肌细胞凋亡指数(AI),免疫组化法检测线粒体融合蛋白2(Mnf2)表达,免疫印迹法检测磷酸化Akt(p-Akt)表达,并进行比较。结果与对照组相比,AMI组及Statin1、Statin2组心肌细胞AI升高,p-Akt表达下降,Mnf2表达增加(P均<0.05);与AMI组相比,Statin1、Statin2组心肌细胞AI和Mnf2表达均显著降低(P均<0.05),而p-Akt表达增高(P<0.05),且Statin2组较Statin1组变化更为显著(P<0.05)。结论 瑞舒伐他汀可抑制大鼠AMI后的细胞凋亡,其作用可能与下调Mnf2的表达有关,且呈一定剂量依赖性。
急性心肌梗死;细胞凋亡;线粒体融合蛋白2; Ras-PI3K/Akt;瑞舒伐他汀;大鼠
急性心肌梗死(AMI)后坏死心肌介导的炎症反应,在促进坏死心肌修复愈合的同时,也加速心肌的重构并通过细胞因子的级联效应导致心肌细胞凋亡。线粒体融合蛋白2(Mfn2)是线粒体外膜上的一种跨膜蛋白,其可能通过激活线粒体凋亡途径,或抑制Ras-磷脂酰肌醇3-激酶(PI3K)-蛋白激酶B (PKB)通路,诱导心肌细胞发生凋亡[1]。他汀类药物除抑制甲羟戊酸合成,还可抑制类异戊二烯的生成,影响Ras-PI3K/Akt通路,发挥其调节细胞凋亡、改善血管内皮功能、抗血小板、调脂、抗动脉粥样硬化等作用[2]。本实验于2015年4~10月建立大鼠AMI模型,研究瑞舒伐他汀对AMI大鼠心肌细胞凋亡的抑制作用,并探讨其作用机制。
1 材料与方法
1.1材料健康成年SPF级SD雄性大鼠60只,体质量250~300 g,购自山西医科大学实验动物中心。试剂及药品:Mfn2兔多克隆抗体(武汉博士德生物工程有限公司);PV9000试剂盒、DAB试剂盒(北京中杉生物工程公司);凋亡试剂盒(德国roche公司);瑞舒伐他汀钙片(10 mg/片),AstraZeneca公司。仪器:日本尼康显微照像系统;微波炉(格兰仕);温箱(电热恒温水浴箱DHG-9031,上海);电子天平(BS224S,德国)。
1.2动物分组及模型建立健康成年SPF级SD雄性大鼠60只,适应性喂养7 d。随机抽取10只为对照组,前降支下穿线不结扎;其余50只制作AMI模型:用10%的水合氯醛3 mL/kg腹腔注射麻醉大鼠,气管切开插管,呼吸机辅助呼吸,潮气量15 mL/kg,呼吸频率70~80次/min,呼吸比2∶1,开胸后分离胸膜、心包,挤出心脏,以医用缝线穿圆形塑料套管前结扎大鼠左前降支阻断血流,造成AMI模型。24 h后存活41只大鼠,按照随机数字表法分为3组,AMI组13只、Statin1组14只、Statin2组14只,术后第1天起Statin1、2组分别给予瑞舒伐他汀1.5、3.0 mg/(kg·d)灌胃,对照组及AMI组均以等量生理盐水灌胃,每3天称重1次并调整给药剂量,共给药4周。实验结束时各组存活量分别为对照组10只、AMI组9只、Statin1组11只、Statin2组13只。
1.3心肌细胞凋亡指数(AI)的测算用转移酶介导脱氧尿苷三磷酸缺口末端标记法(TUNEL)对组织石蜡切片行凋亡染色。凋亡细胞出现棕黄色颗粒为阳性,每张标本取3张切片,每张切片在心梗区域随机取5个高倍镜视野,记录阳性细胞数,计算AI,AI=凋亡细胞核数/总细胞核数×100%。
1.4心肌组织中Mfn2表达的检测采用免疫组化法。处死大鼠后取心脏标本,制作石蜡切片,选取梗死心肌组织边缘区域(对照组在左室前壁)切片。依照试剂盒步骤检测Mfn2。阳性切片作阳性对照,PBS切片作阴性对照,细胞内出现棕黄色颗粒为阳性,每张标本取3张,分析Mfn2蛋白阳性染色指数(阳性细胞数/视野细胞总数×100%)。
1.5心肌组织中p-Akt表达的检测采用免疫印迹法。提取组织总蛋白,测定蛋白浓度。 蛋白上样量为50 μg,经电泳、转膜、封闭后,加入一抗(抗α-actin、抗p-Akt、抗Akt),4 ℃放置12 h以上,加入辣根过氧化物酶偶联的二抗,平稳摇动,室温放置2 h后检测。
2 结果
2.1各组AI、Mfn2比较见表1。
2.2各组心肌组织 p-Akt 的表达比较与对照组比较,AMI组及Statin1、Statin2组心肌组织中p-Akt蛋白表达均降低(P均<0. 01);与AMI组比较,Statin1组、Statin2组p-Akt表达增加(P均<0.05) ,以Statin2组为著(P<0.05) ,见图1、表1。
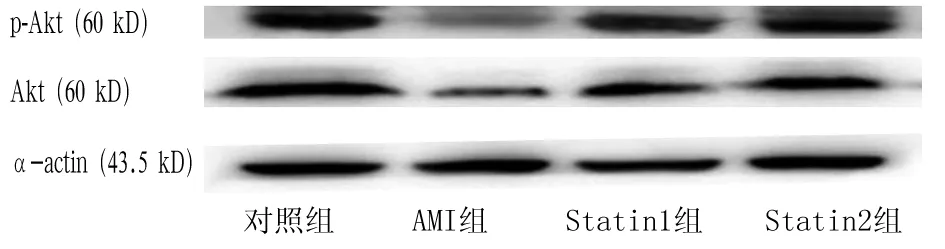
图1 各组p-Akt表达的免疫印迹图
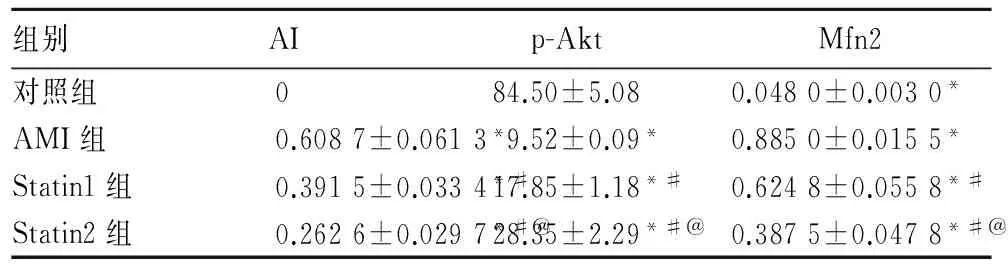
组别AIp-AktMfn2对照组084.50±5.080.0480±0.0030*AMI组0.6087±0.0613*9.52±0.09*0.8850±0.0155*Statin1组0.3915±0.0334*#17.85±1.18*#0.6248±0.0558*#Statin2组0.2626±0.0297*#@28.35±2.29*#@0.3875±0.0478*#@
注:与对照组比较,*P<0.05;与 AMI组比较,#P<0.05; 与Statin1组比较,@P<0.05。
3 讨论
既往认为AMI时以心肌细胞坏死为主。近年来多项相关动物实验证实,AMI后细胞凋亡才是心肌细胞死亡的主要方式[3]。Kajstura等[4]在AMI动物模型中发现,梗死后约6 h心肌细胞凋亡可达到高峰,7 d仍有大量的心肌细胞发生凋亡。Bardales等[5]用TUNEL法检测85例AMI死亡患者尸解的心肌细胞,凋亡率超过90%。
AMI发生后,缺氧、缺血、再灌注等诱因激活各种凋亡因子,调控相关信号通路,诱导细胞凋亡[6]。细胞凋亡信号转导为多途径且相互影响,研究发现,Ras-PI3K/Akt 路径是促使心肌细胞发生凋亡的主要通路[7]。本实验结果显示,AMI组较对照组细胞凋亡明显增多,同时心肌组织中p-Akt表达较对照组也明显降低。以上结果均表明AMI后大量心肌细胞凋亡,与Ras-PI3K/Akt通路激活密切相关。
原癌基因Ras不仅能调控PI3K/Akt 通路,诱导细胞凋亡,还可通过PI3K非依赖型途径直接激活Akt[8]。研究表明,Mfn2在心肌、骨骼肌和脑组织中高表达,其可能参与心肌细胞凋亡的早期活化,加速线粒体分裂,促使线粒体体积减小[9]。Mfn2对Ras有直接负调控作用,能阻滞Ras-PI3K/Akt 通路,抑制Akt磷酸化,继而使B淋巴细胞瘤-2基因表达减少,促凋亡基因Bax表达增加,通过线粒体相关途径参与细胞凋亡[10]。本实验结果显示,大鼠AMI造模成功后,梗死及周围心肌组织Mfn2表达明显增高,p-Akt表达明显降低,这表明梗死后损伤激活Mfn2的表达,弱化Akt 磷酸化从而促发心肌细胞凋亡。
大量的循证医学证实,他汀类药物在冠心病防治过程中占据着不可替代的地位。随着临床广泛应用及研究的深入,人们发现他汀类药物有重要的独立于调脂外的多重作用[11]:他汀类药物除抑制甲羟戊酸合成,阻断Cho的生成过程,还可抑制类异戊二烯,可与一些单体G蛋白(如Ras,Rho等)偶联,增加后者的亲脂性,影响Ras-PI3K/Akt 及Rho/Rho激酶信号通路、RAAS系统和过氧化物酶增殖物激活受体的活性,发挥其抑制心肌细胞凋亡、改善血管内皮功能、调脂、抗动脉粥样硬化等作用,降低心血管疾病的发病率和病死率[2]。本实验结果显示,瑞舒伐他汀可使Mfn2表达明显下调,抑制Ras相关信号通路的活化,减少大鼠AMI后的细胞凋亡,其作用表现出一定的剂量依赖性。
综上所述,笔者认为Ras介导的PI3K/Akt信号通路可能在AMI后心肌细胞凋亡发生发展过程中起重要作用,而瑞舒伐他汀通过下调Mfn2表达,上调p-Akt水平,最终减少AMI后心肌细胞凋亡,防止AMI范围进一步扩大导致心肌细胞坏死,从而对心功能不全及心室重构有一定的治疗作用。
[1] Guo XM, Chen KH, Guo YH, et al. Mitofusion2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway[J].Circ Res, 2007,101(11):1113-1122.
[2] 赵艳,杜冠华,王少华.Rho/Rho激酶信号转导通路与他汀类药物[J].中国新药与临床杂志,2008,10(27):786-790.
[3] Prech M, MarszaIek A, Schroder J, et al. Apoptosis as a mechanism for the eIimination of cardiomyocytes after acute myocardiaI infarction [J]. Am J Cardiol, 2010,105(9):1240-1245.
[4] Kajstura J, Cheng W, Sarangarajan R, et al. Necrotic and apoptotic myocyte cell death in the aging heart of Fischer 344 rats[J]. Am J Physiol, 1996,271(3 Pt 2):1215-1228.
[5] Bardales RH, Hailey LS, Xie SS, et al. In situ apoptosis assay for the detection of early acute myocardial infarction[J]. Am J pathol,1996,149(3):821-829.
[6] 张丽娜,吴星恒.心肌细胞凋亡与PI 3K/Akt信号途径表达及其调控机制的研究[J].南昌大学学报(医学版),2010,50(5):119-121.
[7] Ishihara Y,Shimamoto N. Sulfaphenazole attenuates myocardial cell apoptosis accompanied with cardiac ischemia-reperfusion by suppressing the expression of BimEL and Noxa[J]. Pharmacol Sci, 2012,119(3):251-259.
[8] 袁向飞,陆敏.Ras/MAPK与PI3K/Akt信号转导通路及其相互作用[J].国际检验医学杂志,2006,156(3):4-5.
[9] Parra V, Eisner V, Chiong M, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis[J]. Cardiovasc Res, 2008,77(2):387-397.
[10] Shen T, Zheng M, Cao CM, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis[J]. J Biol Chem, 2007,282(8):23354-23361.
[11] 鄢华,高炜.他汀类药物作用和作用机制的新认识[J].临床内科杂志,2006,23(1):5-7.
汤建民(E-mail:tjmgrx@163.com)
10.3969/j.issn.1002-266X.2016.17.009
R541.4
A
1002-266X(2016)17-0028-03
2015-11-18)
