Pt(II)催化炔基氮丙啶合成环戊烷吡咯机理的密度泛函理论研究
2016-08-08张金生余晓娟
张金生,余晓娟
(贵州师范大学 化学与材料科学学院,贵州 贵阳 550001)
Pt(II)催化炔基氮丙啶合成环戊烷吡咯机理的密度泛函理论研究
张金生,余晓娟
(贵州师范大学 化学与材料科学学院,贵州 贵阳550001)
摘要:用量子化学密度泛函理论B3LYP方法在6-311G(d,p)的计算水平上对PtCl2催化2-炔基-1-氮杂环己烷合成1,4,5,6-四氢化环戊烷吡咯的反应,进行了计算,找到两条主要的反应通道,最优势反应路径包括炔基的活化、氮丙啶的氮与炔基间的环化反应、环异构化、质子转移和催化剂解离5个步骤。PtCl2催化作用的本质在于Pt2+与炔基配位,能降低炔基反键轨道的能级,降低炔基反键轨道与氮丙啶的氮原子孤电子占据轨道LP-(2p)N的能级差,使丙啶氮原子与炔基之间的环化反应势垒下降。
关键词:炔基氮丙啶;四氢化环戊烷吡咯;环化反应;σ-π配键;密度泛函理论
0引言
环戊烷吡咯是所有叶绿素中发光基团的重要组成部分[1],它可作为合成生物活性分子的中间体[2],人工合成这类化合物具有重要的意义。近年来的实验研究表明,用某些过渡金属催化丙炔氮杂环丙烷,可有效合成多取代吡咯[3],如金(Au)催化炔基氮杂环丙烷合成氮-甲酰吡咯[4],催化α-乙酰炔基环氧乙烷或氮杂环丙烷合成呋喃和吡咯[5],催化炔基氮杂环丙烷合成2,5-双取代吡咯[6]等,铂(Pt)催化环化丙炔环氧乙烷和氮杂环丙烷合成多取代呋喃和吡咯[7],在单质碘作用下环异构化丙炔氮杂环丙烷合成3-碘代吡咯[8]。利用这些人工合成方法,反应能在比较温和的条件下顺利进行,而且产率较高,反应也符合原子经济性原则。
在2011年,Masahiro Y[9]等报道,在加热至373K的条件下,PtCl2能催化2-炔基-1-氮杂环己烷,合成1,4,5,6-四氢化环戊烷吡咯,如图1所示。R1、R2、R3和溶剂的不同,将会得到不同的产率。以二噁烷为溶剂,R1=C3H7、R2=H、R3=H时,产率可高达97%。有趣的是,在该反应中,丙啶氮原子与炔基一位碳原子C(1)结合成环,C(5)与螺原子C(4)之间的σ键断裂,与C(3)生成σ键,从而由原来的四元环结构变成了五元环结构。

图1 PtCl2催化炔基氮丙啶合成四氢化环戊烷吡咯Fig.1 Tetrahydrocyclopenta[b]pyrrole synthesis from 1-benzyl-2-ethynyl-1-azaspiro [2.3] hexane catalyzed by PtCl2
为了探索这个反应的机理,揭示催化剂PtCl2的催化本质,我们用量子化学密度泛函理论,对图1中的R1=C3H7、R2=H、R3=H的反应体系进行了研究。
1计算方法和模型
用量子化学密度泛函理论[10]B3LYP方法对如上反应体系进行计算。对Pt原子采用LANL2DZ基组,并增加一套f轨道的极化函数,其极化系数为0.993[11],对其它原子均采用分裂基组6-311G(d,p)的计算水平,对反应势能面上所有驻点的几何结构进行了全优化。在相同的计算水平上,对所有优化结构进行了振动频率计算,获得了所有优化结构的热力学数据、振动频率(ν)和零点振动能,并对所有过渡态进行了内禀反应坐标的计算,以确认过渡态的真实性。用自洽反应场极化连续介质模型(PCM)计算了各结构在溶剂二噁烷中的溶剂化效应能,用自然键轨道理论对反应中的重要结构进行了自然键轨道分析。所有计算均由Gaussian03程序完成。
2结果与讨论
计算结果表明,反应物1、产物2、催化剂PtCl2和反应过程中的所有中间体的力常数本征值全是正值,并对它们进行了波函数稳定性测试,说明它们是反应势能面上的稳定点。反应路径中的所有过渡态均有且只有一个虚频,用GView3.07程序查看该频率的振动模式,发现其振动方向与化学键的形成或断裂的方向一致,而且这些过渡态的内禀反应坐标的计算,也确认了它们是连接其前、后稳定结构的鞍点。对PtCl2催化2-炔基-1-氮杂环己烷以合成1,4,5,6-四氢化环戊烷吡咯的反应,找到了2条主要通道,如图2所示。反应中各驻点的几何结构和反应势能面示意图分别如图3、图4所示,各驻点的热力学数据和主要自然键轨道能级比较分别列于表1、表2。
在第一条反应路径中,PtCl2的铂原子与反应底物1的炔基π键配位,生成中间体IM1。如表1所示, 该配位反应的吉布斯自由能变为-151.7 kJ·mol-1,放热176.3 kJ·mol-1。
由于PtCl2催化剂与炔基配位,使IM1的电子结构与反应底物1有如下3个显著不同之处,从而使反应底物活化。
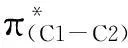
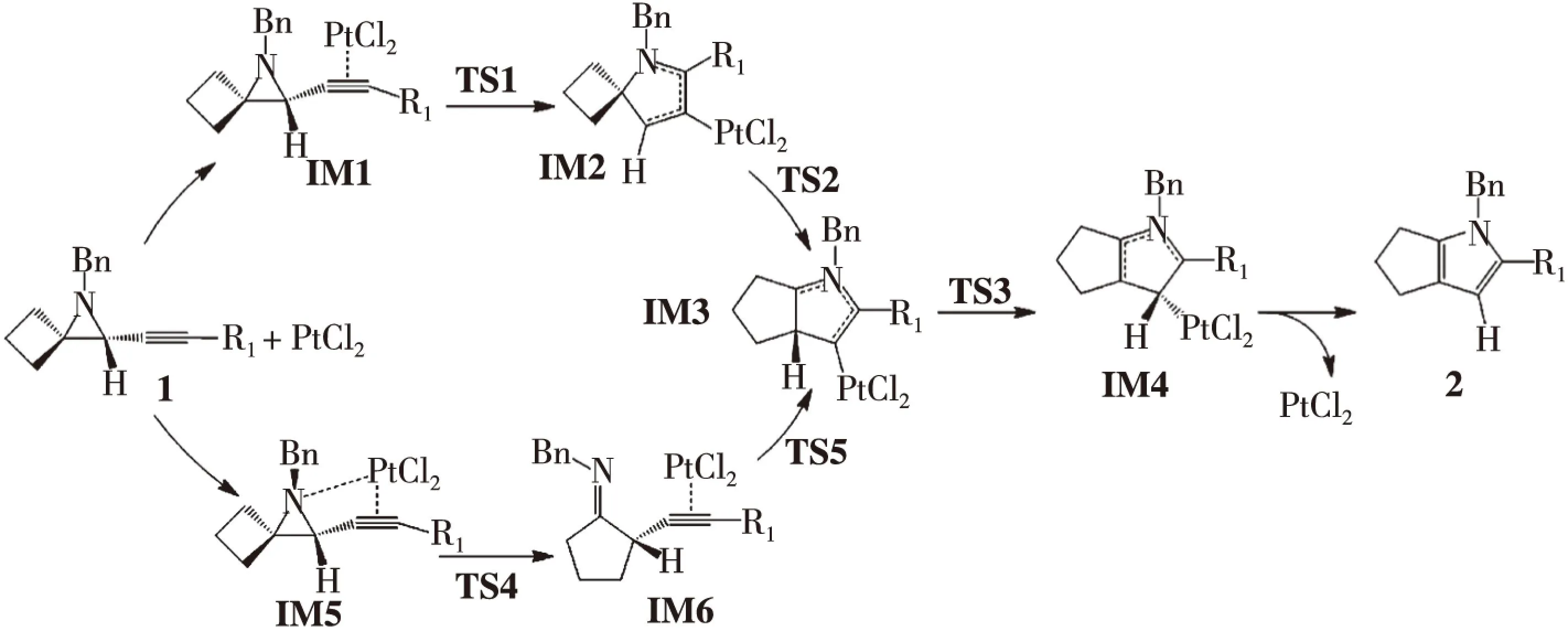
图2 PtCl2催化炔基氮丙啶合成四氢化环戊烷吡咯反应的可能路径Fig.2 The reaction channels of PtCl2-catalyzed 1-benzyl-2-ethynyl-1-azaspiro [2.3] hexane to synthesize tetrahydrocyclopenta [b] pyrrole
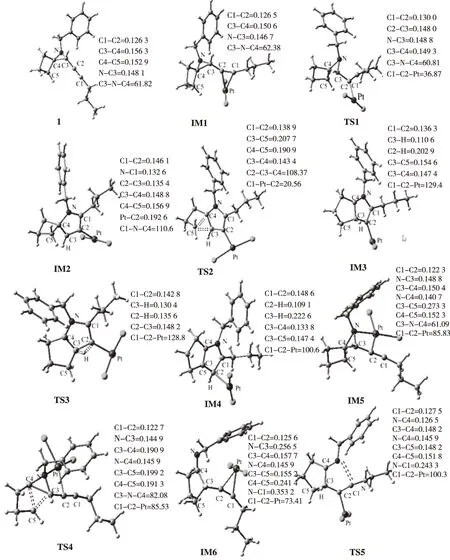
图3 B3LYP/6-311++G(d,p)优化的驻点结构(键长:nm,键角:o)Fig.3 Geometries of the stationary points optimized at B3LYP/6-311++G** calculation level (bond lengths in nm and bond angles in degree)
第二,Pt2+与炔基配位,还使丙啶氮原子极化张量(APT)正电荷增加(在1和IM1中分别为+0.330和+0.445),也使C(1)的APT负电荷增加(在1和IM1中分别为-0.635和-0.817)。这意味着氮丙啶氮原子的亲核性增强了,丙啶氮原子与C(1)之间的静电作用也增强了,氮丙啶的氮与炔基碳发生亲核环化反应更容易进行。
IM1中氮丙啶的氮原子进攻炔基中的C(1)时,即发生分子内环化反应,经过渡态TS1,生成氮杂环化合物IM2。在此过程中,氮丙啶的氮原子作为电子受体,炔基作为电子受体。在氮原子进攻C(1)的同时,C(1)离开Pt原子,而C(2)接近Pt原子。TS1是具有早期势垒的紧凑过渡态,正向反应势垒ΔG≠=60.0 kJ·mol-1。在过渡态TS1中,σ(C1-N)的逐渐形成,C1≡C2转变成双键,Wiberg键级为1.13(见表2),Pt与C(2)形成σ键。
IM2可通过σ(C4-C5)键的断裂和σ(C3-C5)键的形成,经过渡态TS2,形成具有两个五元环的中间体IM3。TS2是具有早期势垒的过渡态,ΔG≠=90.8 kJ·mol-1。在IM2中,C(3)和C(5)的原子轨道都接近sp3杂化。C(3) 和C(5)的原子极化张量(APT)电荷分别为-0.054和0.063,这有利于σ(C3-C5)的形成。TS2中,化学键σ(C4-C5)明显削弱,C(3)和C(5)间Wiberg键级为0.3518,表明σ(C3-C5)键在形成。IM3中,化学键σ(C3-C5)已经形成,σ(C3-C5)= 0.686(d2.87s)C5+0.727(sp2.73)C3,Wiberg键级为0.9647,电子占据数为1.948e。
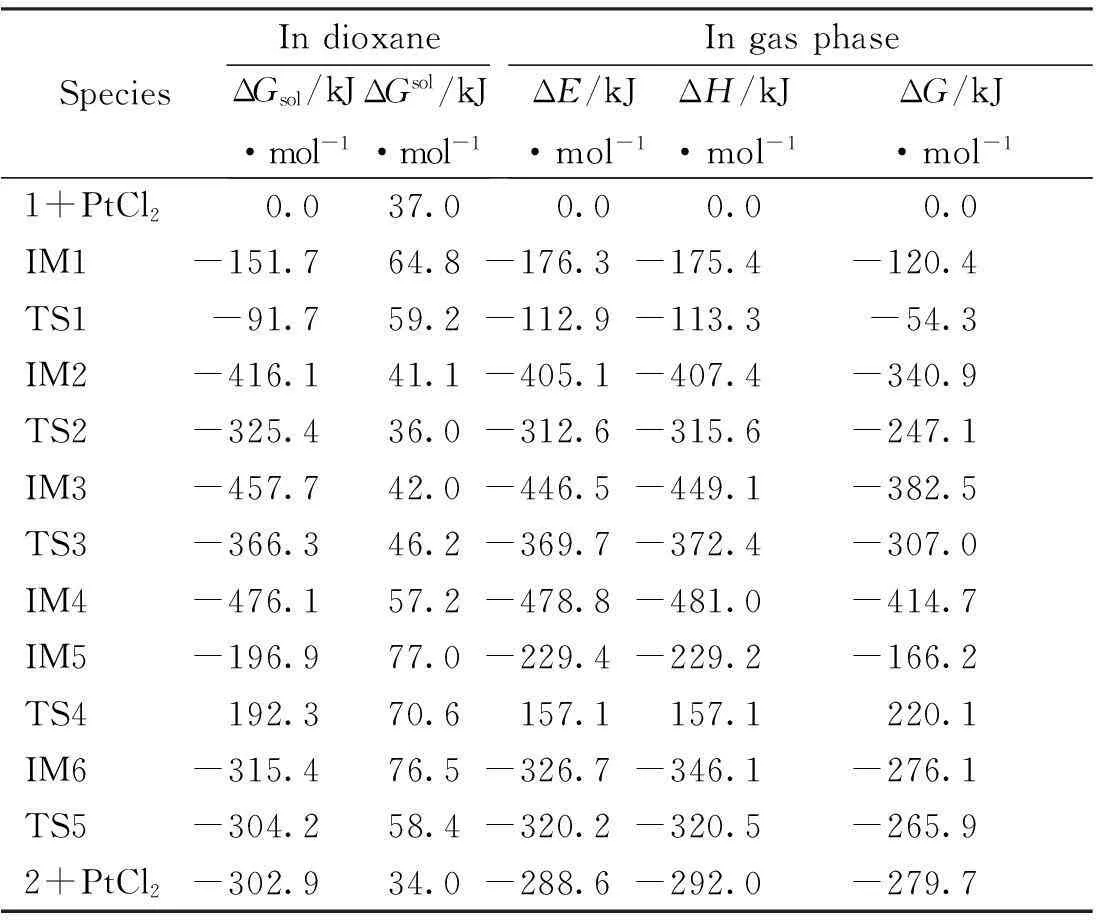
表1 各驻点在溶剂二噁烷中的相对自由能ΔGsol,溶剂化效应能ΔGsol, 以及气相时的相对能ΔE、相对焓ΔH和相对自由能ΔG

表2 对反应驻点1,IM1,TS1,IM5,TS4和IM6的相关自然键轨道能级ε(kJ·mol-1)比较
中间体IM3可以发生质子转移反应,C(3)上的H直接转移到C(2)上,经过渡态TS3,生成中间体IM4。TS3是具有早期势垒的松散过渡态,正向反应势垒ΔG≠=91.4 kJ·mol-1。最后中间体IM4解离出催化剂PtCl2,得到产物1,4,5,6-四氢化环戊烷吡咯2。
在第(2)条反应路径中,PtCl2的Pt2+同时与反应底物1的氮原子和炔基配位,形成中间体IM5。该反应放热229.4kJ·mol-1,其吉布斯自由能变为-196.9kJ·mol-1。由于IM5的三元环和四元环都具有较大的角张力,可通过σ(C4-C3)键、σ(C4-C5)键和σ(N-C3)键的断裂,σ(C3-C5)键的形成,经过渡态TS4,生成中间体IM6。该过程放热97.3kJ·mol-1,正向反应势垒ΔG≠=389.2 kJ·mol-1。

表3 主要结构中的部分原子的APT电荷δ和部分化学键键级Pij
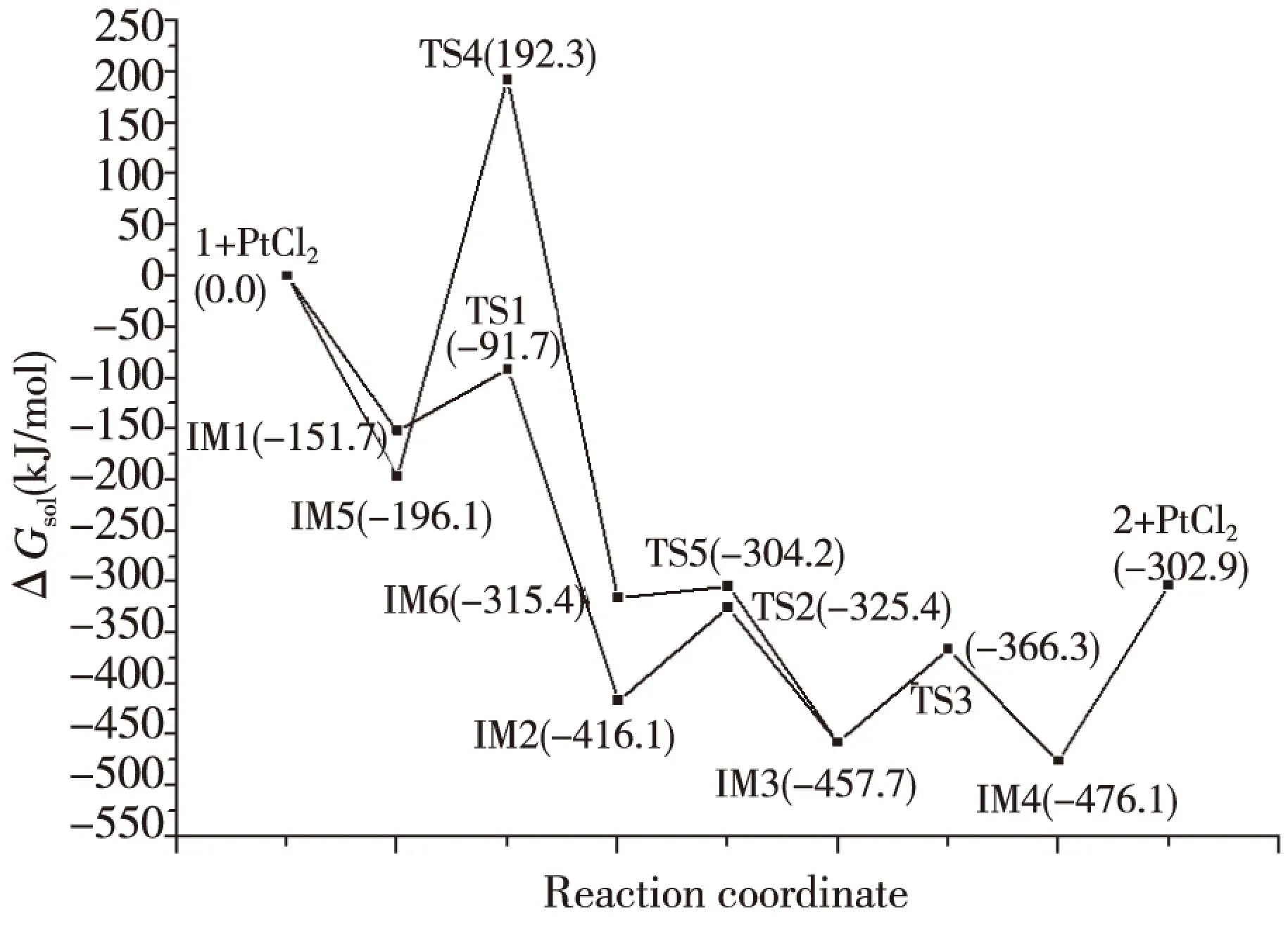
图4 PtCl2催化炔基氮丙啶合成四氢化环戊烷吡咯的反应势能面Fig.4 PdCl2 catalytic alkynyl aziridine synthesis tetrahydronaphthalene cyclopentane pyrrole reaction potential energy surface
IM6的氮原子进攻炔基中的C(1)时,可发生环化反应,经过渡态TS5,生成氮杂环化合物IM3。在该步骤中,氮原子作为电子给予体,炔基作为电子受体。在氮原子靠近C(1)原子的同时,C(1)原子远离Pt2+,而C(2)接近Pt2+。该反应放热119.8kJ·mol-1,势垒ΔG≠=11.2kJ·mol-1。
随后与第(1)条反应通道一样,IM3经过渡态TS3,生成中间体IM4,再解离出催化剂PtCl2,得到最终产物2。
反应通道(1)与(2)的不同之处在于催化剂PtCl2与反应底物1的配位方式不同。前者的Pt2+与炔基的C1≡C2配位,后者的Pt2+在与炔基的C1≡C2配位的同时,还与氮丙啶的氮原子配位。在1→IM3的2条反应路径中,由于TS4比TS1的势垒高出328.4kJ·mol-1,因此反应通道(1)在动力学上占优势,是优势反应通道。
3结论
参考文献:
[1] NAKAMURA H,KISHI Y,SHIMONURA O,et al.Structure of dinoflagellate luciferin and its enzymic and nonenzymic air-oxidation products [J].J Am Chem Soc,1989,111(19):7607-7611.
[2] FÜRSTNER A.Chemistry and biology of roseophilin and the prodigiosin alkaloids: A survey of the last 2500 years [J].Angew Chem Int Ed,2003,42(31):3582-3603.
[3] CHEN D D,HOU X L,DAI L X.A facile and regioselective synthesis of 2,5-disubstituted pyrroles via gold-catalyzed cycloisomerization of acetylenylaziridines [J].Tetrahedron Lett,2009,50(50):6944-6946.
[4] DU X,XIE X,LIU Y.Gold-catalyzed cyclization of alkynylaziridines as an efficient approach toward functionalized N-Phth pyrroles [J].J Org Chem,2009,75(2):510-513.
[5] BLANC A,ALIX A,WWIBEL J,et al.Gold(I)-catalyzed tandem rearrangement -nucleophilic substitution of α-acetoxy alkynyl oxiranes or aziridines: efficient approach to furans and pyrroles [J].Eur J Org Chem,2010,2010(9):1644-1647.
[6] DAVIES P W,MARTIN N.Aryl-substituted N-tosyl alkynyl aziridines undergo a gold-catalyzed ring expansion to afford 2,5-substituted pyrrole products. depending on the counterion to the gold catalyst and the solvent, a Ring-expansion and rearrangement leads to 2,4-substituted pyrroles[J].Org Lett,2009,11:2293-2296.
[7] YOSHIDA M,AL-AMIN M,SHISHIDO K.Highly substituted furans were conveniently synthesized by the platinum-catalyzed reaction of propargylic oxiranes.Propargylic aziridines were also reacted with the platinum catalyst to produce the corresponding substituted pyrroles in good yields [J].Synthesis,2009:2454-2466.
[8] YOSHIDA M,EASMIN S,AL-AMINI M,et al.Synthesis of substituted 3-iodopyrroles by cycloisomerization of propargylic aziridines with iodine[J].Tetrahedron,2011,67(18):3194-3200.
[9] YOSHIDA M,MAEYAMA Y,AL-AMIN M,et al.Synthesis of substituted 1,4,5,6- tetrahydrocyclopenta[b]pyrroles by platinum-catalyzed cascade cyclization/ring expansion of 2-alkynyl-1-azaspiro[2.3]hexanes [J].J Org Chem,2011,76(14):5813-5820.
[10]PARR R G,YANG W.Density-functional theory of atoms and molecules [M].New York:Oxford University Press,1989:217-219.
[11] EHLERS A W,BÖHME M,DAPPRICH S,et al.A set of f-polarization functions for pseudo potential basis sets of the transition metals Sc-Cu,Y-Ag and La-Au [J].Chemical Physics Letters,1993,208(1):111-114.
文章编号:1004—5570(2016)01-0055-06
收稿日期:2015-10-31
基金项目:国家自然科学基金(No. 21163003)
作者简介:张金生(1970-),男,博士,教授,研究方向:应用量子化学研究,E-mail: zjs-xs@163.com.
中图分类号:O641-3
文献标识码:A
DFT study on the PtII-catalyzed tetrahydrocyclopenta[b]pyrroles synthesis from 2-alkynyl-1-azaspiro[2.3]hexanes
ZHANG Jinsheng,YU Xiaojuan
(School of Chemistry and Materials Science, Guizhou Normal University, Guiyang, Guizhou 550001, China)
Abstract:By means of density functional theory (DFT), the PtII-Catalyzed mechanism for 1-benzyl-1,4,5,6-tetrahydrocyclopenta[b]pyrrole synthesis from 1-benzyl-2-ethynyl-1-azaspiro[2.3]hexane was investigated. Two reaction channels were found. The overall reaction in the favored channel includes the activation of alkynyl, cyclization reaction, isomerization reaction, proton migration and regeneration of the catalyst. The substrate 1 was activated by its combination of Pt(II) to triple bond which leads to a decrease in the orbital energy of π*(C1-C2)and the orbital energy-gap between π*(C1-C2)and LP-(2p)N.
Key words:alkynyl-1-azaspiro[2.3]hexanes; tetrahydrocyclopenta[b]pyrroles; cyclization reaction; σ-π coordination bond; density functional theory
