“受阻Lewis酸碱对”化学的研究进展
2016-07-27柳晗宇杨中天余子迪米天雄北京大学化学与分子工程学院北京100871
柳晗宇 杨中天 余子迪 米天雄 卞 江(北京大学化学与分子工程学院,北京100871)
·今日化学·
“受阻Lewis酸碱对”化学的研究进展
柳晗宇 杨中天 余子迪 米天雄 卞 江*
(北京大学化学与分子工程学院,北京100871)
摘要:受阻Lewis酸碱对(Frustrated Lewis Pairs,FLPs)是一类具有特殊反应活性的Lewis酸碱对。自发现以来,FLPs受到了广泛关注并在许多领域崭露头角。本文对FLPs在不对称氢化、高分子聚合、CO2催化还原等应用领域取得的突破进行了介绍;同时对过渡金属FLPs和FLPs配位的过渡金属催化体系进行了综述;最后对FLPs领域未来的发展前景进行了展望。
关键词:受阻Lewis酸碱对;不对称氢化;离子型聚合反应;CO2催化还原;过渡金属催化体系
1923年,Lewis[1]提出了按照电子给受体分类的Lewis酸碱体系,极大地拓展了酸碱概念的内涵,Lewis酸碱体系也成为了无机、有机和配位化学中最基本的概念之一。长期以来,作为反应产物的Lewis酸碱加合物一直被认为是热力学惰性的“能量尽头”,并没有得到足够的重视。然而,某些具有特殊结构的Lewis酸碱组合可以共存于体系中,并具有潜在的小分子活化性能。
2006年,Stephan小组[2]将常见的Lewis酸(B(C6F5)3)与Lewis碱(PHMes2)混合,发现二者并没有发生典型的加合反应,而是发生取代生成化合物2(图1)。令人振奋的是,2经Me2SiHCl还原,竟得到了同时带有正、负电性氢的稳定的化合物3。更进一步,3在100°C以上释放H2生成化合物4;而在室温下4也可以快速、自发地与H2生成加成产物3。3、4的存在,为室温下氢气的非金属活化提出了可能性。
化合物4为什么对H2有着特殊的活化能力?究其原因,这种反应活性来源于其所带的大位阻基团,这些基团的存在使得4中的酸碱位点不能发生分子间配位反应。这样,保留下来的自由酸碱位点可以与H2作用,使其异裂生成活化产物3。事实上,后续发现的一系列酸碱组合,如P(t-Bu)3/B(C6F5)3、(i-Pr)2NH/B(C6F5)3等,也可以使H2发生异裂活化,生成带有正、负电性氢的离子对。Stephan将这种位阻极大的Lewis酸碱组合称为“受阻Lewis酸碱对”(Frustrated Lewis Pairs,FLPs)。英语中frustrated的意思是“失意的,沮丧的,挫败的”,国内译成“失配”或“受阻”,这个词很好地体现了大空阻Lewis酸碱间无法形成加合物的状态。这些看似失意的Lewis酸碱组合却有着令人激动的反应性能,为H2、CO2、N2O、烯烃、炔烃等一系列小分子的活化打开了大门[3,4]。自发现以来,FLPs迅速成为热门的研究领域,经过10年的发展,FLPs已经在诸多催化领域中展现了优于传统催化剂的一系列特性,为不对称氢化、高分子聚合等领域提供了新的发展机遇。
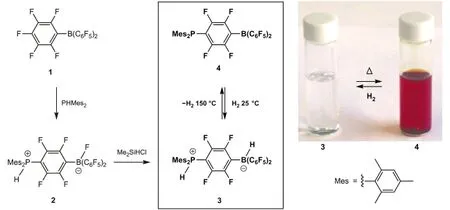
图1 FLPs对氢气的活化
1 FLPs催化的不对称氢化反应
不对称反应是引入手性中心的关键反应,不对称氢化反应是其中极其重要的一类,其中以氢气为氢源的非金属催化的不对称氢化反应具有很大优势,受到了广泛重视,并取得了一系列成果。最初的FLPs催化不对称氢化案例来源于手性底物的诱导,比如Soós小组[5]以MesB(C6F5)2/DABCO为催化剂对D-香芹酮进行的氢化(图2)以及Stephan小组[6]和Klankermayer小组[7]报道的B(C6F5)3催化手性亚胺的氢化;进一步的研究扩展到手性的Lewis酸或Lewis碱催化不对称氢化反应,一般来说,后者具有更为广阔的应用空间。
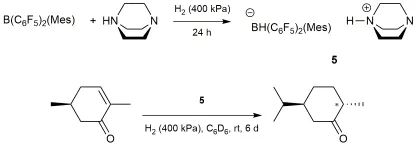
图2 FLPs活化H2及后续加成
自2008年Klankermayer小组首次报道了手性FLPs的不对称氢化反应后,Klankermayer小组[8]、Repo小组[9]、Stephan小组[10]和中国的杜海峰教授小组[11]等先后开发了不同的手性Lewis酸或Lewis碱催化剂(图3),以期提高反应的活性和对映选择性并简化操作步骤。

图3 业已报道的手性FLPs催化剂
FLPs催化不对称氢化是一个崭新的研究领域,为科研工作者带来了新的研究思路。在目前的研究工作中,FLPs已经展现出优于传统催化剂的化学选择性和立体选择性,并在不对称催化中展现出较好的不对称选择性。
与此同时,作为一个刚刚起步的研究领域,FLPs催化不对称氢化仍面临很多挑战,比如由于研究的课题组比较少,有效的催化剂体系极少,底物范围比较窄,官能团耐受性不高。结合计算化学的手段,开发新颖、高效的催化剂,进一步提高氢化反应的效率和选择性,拓展底物适用范围是这一领域未来的重点研究方向。
2 FLPs作为高分子聚合催化剂
FLPs在其相当短的发展历史里已在化学的诸多方面取得了非常突出和卓越的进展,其中也包括了高分子化学。材料性能需求的不断提升,要求化学工作者们开发新的聚合单体和催化剂。Lewis酸碱对以其独特的协同活化作用(图4),为离子型聚合反应提供了新的思路,引起了化学工作者的广泛关注[12]。
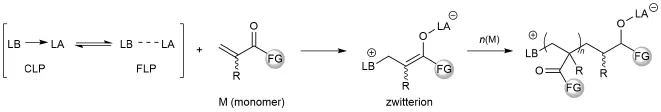
图4 Lewis酸碱对催化的高分子聚合反应
2.1 CLPs催化高分子聚合:历史与局限性
经典Lewis酸碱对(Classical Lewis Pairs,CLPs)作为高分子聚合的催化剂已经有不短的历史,1960年,Murahashi等[13]报道了2:1的Et3Al和PEt3可以引发甲基丙烯酸甲酯(MMA)聚合,得到高度间同聚合的产物。1971年,Ikeda等[14]研究了基于AlEt3、InEt3等酸和PPh3等碱组成的经典Lewis酸碱对催化丙烯腈和甲基丙烯酸甲酯的聚合。1992年,Kitayama等[15]又重新研究了AlEt3/PR3催化的甲基丙烯酸酯的低温聚合反应(-78/-93°C)。这些研究发现:
(1)单独的Lewis酸或碱均不能引发聚合反应;
(2)催化剂活性不高,高负载下反应时间也较长,TOF值较低;
(3)反应机理不明确。
CLPs在反应中的局限性来源于Lewis酸碱很强的亲和性,导致活化产物不稳定;而FLPs的发现,恰恰为这类问题的解决打开了一扇窗口。2011年,大空阻强酸Al(C6F5)3被应用于MMA的聚合反应,得到了比传统Lewis酸更加优异的结果[16]。由此,FLPs在高分子聚合中的应用受到了高度的关注。
2.2 主族FLPs催化高分子聚合
在其优异的反应性能被发现之后,Al(C6F5)3对各种单体的催化反应被系统地研究。从最早的MMA开始,Zhang等[17]研究了12种结构各异单体的催化聚合反应。
前文已讨论过,单独的Lewis酸或碱不能催化聚合反应,但是值得注意的是,两种高极性的氮杂卡宾Lewis碱TPT和t-BuI可以一定程度上催化MMA聚合。Al(C6F5)3/LB催化体系对于可再生的单体MBL和γ-MMBL的聚合均具有极好的催化效果,而对于环状内酯的开环聚合则效果欠佳,值得进一步研究。在具体操作时,无单体的情况下混合Lewis酸与碱会造成失败的反应结果,而先混合Lewis酸与单体,令其反应形成加合物,再加入Lewis碱则能得到满意的结果。在将Al(C6F5)3替换为酸性较弱的高位阻Lewis酸,如ClAl(C6F5)2、i-Pr3Si+、B(C6F5)3时,研究者发现大部分尝试未收到较好的效果。这反映了Lewis酸的高位阻和强酸性在聚合反应中缺一不可[18]。
2.3 活性中间体表征和机理研究
位阻大,酸性较强的FLPs可以稳定聚合反应的中间体,从而进行谱学表征。MMA-Al(C6F5)3和P(t-Bu)3在室温下反应生成的中间体(t-Bu)3PCH2C(Me)=C(OMe)O―Al(C6F5)3(Z/E=3:7)已经成功被核磁共振光谱所表征[19]。NMR数据否认了去质子化后的单独的季鏻盐存在。中间体结构已经由X射线衍射测定,给出的键长数据如Al―O键长(0.17513(11)nm),确证了中间体是两性离子而非膦(图5)。

图5 MMA-Al(C6F5)3/P(t-Bu)3两性离子结构
中间体的表征为机理研究做了铺垫,用计算化学的手段研究了两性离子的形成以及接下来的链引发和链增长过程。采用BP86算法计算了Al(C6F5)3/P(t-Bu)3、PMes3、PPh3和IMes酸碱对催化MMA聚合的相关过程。由MMA-LA加合物产生两性离子的过程如图6所示。两性离子的能量强烈依赖于碱的强度,PMes3对应的能量极高,与其不能催化聚合的实验事实相符。
图7对比了链引发过程两种可能的机理——单金属(unimetallic)和双金属(bimetallic)机理。由能量对比不难看出,无论是何种LB参与反应,聚合都强烈倾向于采取双金属机理。实验事实表明,MMA的聚合速率与[Al]/LB呈现较好的正相关,与计算分析的结果一致。

图6 不同LB形成的两性离子的能量(单位:kJ·mol-1)
虽然FLPs催化的高分子聚合反应发现时间不长,但是已经展现出诱人的发展前景。现阶段,我们对其催化的立体选择性、催化剂的范围、催化剂的活性控制等方面了解还不够深入。开发价格更加低廉、底物适用范围更广、立体选择性更高、聚合速率更快的高分子聚合催化剂是当前研究者的目标。过渡金属FLPs的引入以及计算化学对催化剂设计和机理研究的应用,也为研究者们提供了新的研究方向与机遇。

图7 链引发的两种机理对比
3 FLPs催化的CO2还原
随着化石燃料的迅速衰退,人们将目光投向了大气中丰富的CO2资源,可再生碳基燃料开发与再利用成为了业界关注的重点。虽然目前可以通过水煤气逆反应与Fischer-Tropsch反应[20]固定CO2,但其苛刻的条件、受限的选择性与较低的当量转化率难以令人满意。因而定向、均相、温和的CO2还原反应的开发受到化学工作者的高度重视,FLPs对CO2优良的活化作用恰好对这类难题提供了一种解决方案。
3.1 CO2的活化:奠基与演化
第一个FLPs参与的CO2活化反应由Stephan与Erker小组[21]于2009年报道。他们发现经典的双分子FLPs可以在溴苯溶液中与CO2快速反应生成加合物(图8)。然而,虽然许多FLPs都可以与CO2反应,但由于生成的加合物稳定性较差(大多数在-20°C分解),CO2的进一步还原变得异常困难。
为了增强加合物的稳定性,Stephan小组[22]使用了偕二硼烷化合物Me2C=C(BR2)2(R=Cl,C6F5)代替原有的Lewis酸B(C6F5)3,生成的加合物可以在15°C时稳定存在,其结构为一离域负离子(图9)。另一种思路是使用更加活泼的FLPs,增加正反应的倾向性,如利用碱性更强的TMP(2,2,6,6-四甲基哌啶)/B(C6F5)3与CO2反应,生成的化合物在110°C下仍不分解[23]。
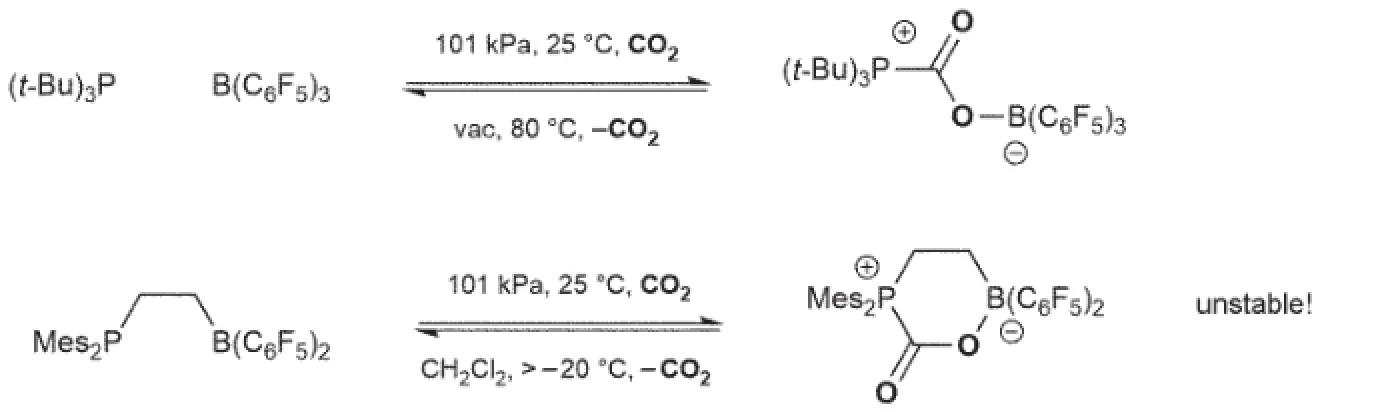
图8 最早发现的FLPs CO2活化反应
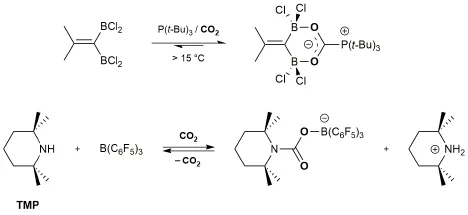
图9 两种使加合物稳定的方案
3.2 CO2的催化还原:突破与改进
前人对FLPs活化反应的不断发现与讨论,使得CO2催化还原体系的设计成为可能。2009年,在Sumerin等[24]报道的B(C6F5)3/TMP分别独立活化H2和CO2的基础上,Ashley与O′Hare小组[22]在100°C时将CO2与B(C6F5)3/TMP活化H2的产物[TMPH][HB(C6F5)3]反应,突破性地得到了CO2还原产物[TMPH][HCO2B(C6F5)3](6)。进一步将6与H2反应得CH3OB(C6F5)2,经水解最终生成CH3OH,如图10所示,这就是第一个非金属均相还原CO2至CH3OH的反应。令人遗憾的是,这个反应中的FLPs并不能形成催化循环,因为HCO2H的脱氧还原过程中,稳定的B―O键会取代B(C6F5)3中的B―C键,破坏B(C6F5)3生成(C6F5BO)3与C6F5H。
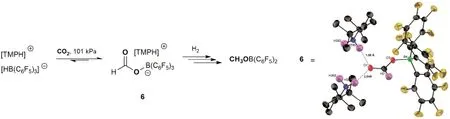
图10 CO2的非金属均相还原
为了使得还原反应能够循环进行,2010年,Piers小组[25]使用还原剂Et3SiH代替H2,利用Si的亲氧能力保护B(C6F5)3不被破坏,得到了较好的产率与较高的TON。除Et3SiH外,9-BBN、BH3NH3[26,27]等含有活泼氢的试剂都可以作为氢的来源。其中比较有趣的发现来自于Stephan小组[28],2013年,Stephan等发现氮杂卡宾与9-BBN形成的加合物可以发生重排,产物恰好可以催化还原CO2(图11)。
虽然利用硅烷、硼烷类试剂可以使得催化循环有效进行,但是由于它们本身的合成需要大量耗能,因而直接使用它们做还原剂是不经济、不现实的。H2作还原剂的FLPs-CO2催化还原仍然是业内研究者的主要目标。不断自我创新、自我完善的FLPs体系为我们进一步研究温和、均相、定向的CO2还原反应的设计提供了大量经验,并指引着我们向更实用的设计不断努力。
4 FLPs与过渡金属:向更高维度发展
FLPs化学的多样性,不仅仅体现为其在化学各领域中的广泛应用(如前文所述),更体现在FLPs本身与化学其他分支的融合再生。虽然FLPs的早期发展立足于对主族元素间酸碱关系的研究,但随着FLPs领域的不断发展,越来越多的工作者将目光投向了周期表的其他疆域,其中过渡金属理所应当地成为了关注的焦点。对于FLPs与过渡金属的研究主要分为两个层面,一层重点研究FLPs作为两性配体与传统过渡金属之间特殊的协同作用,另一层则直接针对以过渡金属作为Lewis酸/碱核心的FLPs进行研究——也就是对“过渡金属FLPs”的研究。
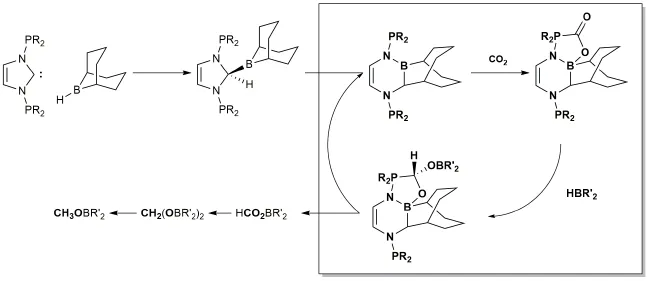
图11 卡宾/BBN重排母体催化还原CO2
4.1 作为两性配体的FLPs
早在2008年,Breher小组[29]就以P/B FLPs体系与过渡金属配合物反应,使FLPs中的Lewis碱与过渡金属发生配位。在随后的几年中,这种“FLPs配体-过渡金属核心”的二元催化体系迅速发展,研究者们不仅开发出了结构各异的配体,更对FLPs配体与过渡金属的各种配位模式进行了积极的探索(图12)。其中,Lewis酸碱位点与过渡金属的双向配位模式引起了业界广泛的关注。
在对于各种配体开发的过程中,双磷氮硼(diphosphine borane,DPB)FLPs配体展示了良好的反应性能。2012年,Harman和Peters[30]将DPB FLPs从传统的FLPs-Pt、Pd、Rh配合物发展到了FLPs-Ni体系,生成的配合物7可实现对H2的活化(图13)。更令人瞩目的是,配合物7甚至实现了对苯乙烯的催化氢化。

图12 FLPs作为配体的配合物体系
在近期的工作中[31],与化合物7具有类似结构的FLPs 8(图14)被证实也具有活化CO、烯烃等小分子的性质,这进一步证实了0价的金属配合物充当了FLPs中的Lewis碱。这项研究为过渡金属CO和CN的催化插入化学研究提供了新的思路。
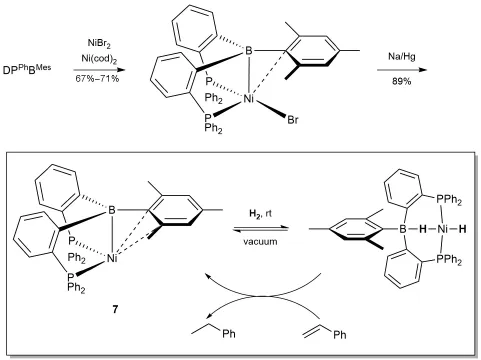
图13 Ni的DPPhBMes配合物的合成及与氢气和苯乙烯的反应
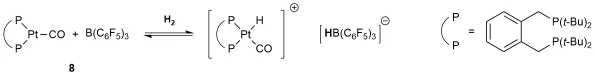
图14 Pt(0)-FLPs对H2的活化
4.2 过渡金属FLPs化合物
如前所述,“过渡金属FLPs”特指过渡金属直接作为Lewis酸/碱核心的FLPs化合物。在对过渡金属FLPs的研究中,最先获得突破的是前过渡金属FLPs,如图15所示,Cp2Zr和(t-Bu)3P对一系列小分子(如N2O、H2、CO2、卤代烃、烯、炔等)均显示出良好的活化作用[32]。
正如主族元素FLPs开发的历程一样,对过渡金属FLPs的研究不仅仅局限在从小分子活化,我们更加期待FLPs可以实现一个完整的催化循环。而这样的突破,最早也是在前过渡金属中实现的。一个十分具有代表性的例子便是对硼胺衍生物脱氢反应:过渡金属FLPs对硼胺衍生物的活化和H2的释放形成了一个完整的催化循环(图16)[33]。
既然前过渡金属FLPs可以形成催化循环,那么天然具有优异催化性能的后过渡金属是否也可以如此?答案是肯定的:2010年,Milstein小组[34]报道了Ru-FLPs的催化反应,配合物9可以催化醇和胺(醇)生成酰胺(酯),而配合物10则可以有选择性地催化生成亚胺(图17)。利用芳构化作用促进FLPs对醇的活化是该反应的一大亮点。

图15 锆的茂化合物和三叔丁基膦对N2O的活化
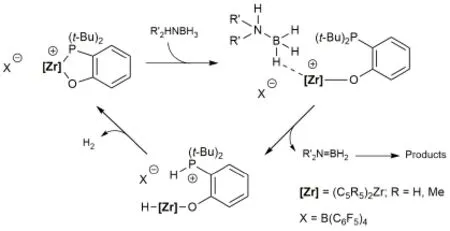
图16 锆FLPs对硼胺衍生物的催化脱氢
综上所述,过渡金属FLPs的早期研究集中在前过渡金属,很大程度上是因为前过渡金属与第三主族元素具有较强的化学相似性——这一点可以从它们类似的离子半径、绝对硬度以及轨道能级上得以解读——导致了前过渡金属FLPs的反应性能与13/15族FLPs相似却又不同。另一方面,具有更多d电子,反应活性更高的后过渡金属FLPs也逐渐进入了大家的视野,成为了新的研究重点。站在更高的层次上,在本章首先介绍的作为配体的FLPs,虽然在形式上与过渡金属FLPs不尽相同,但在本质上,FLPs中的Lewis碱为过渡金属中心提供了电子,而富电子的过渡金属与Lewis酸位点也有相互作用——这本身就是一对过渡金属FLPs的组合。其中的金属甚至同时具有了酸碱反应活性。因此可以预见:在不久的将来,过渡金属与FLPs融合必将达到一个全新的高度,为传统的过渡金属催化反应打开了新的窗口。
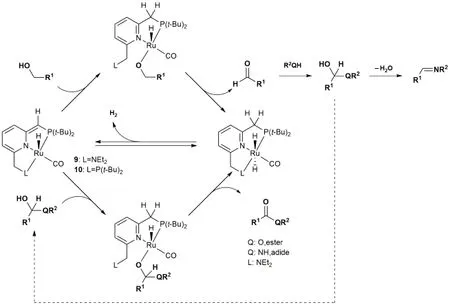
图17 Milstein等[35,36]提出的机理
5 总结与展望
自2006年Stephan小组发现并提出“受阻Lewis酸碱对”这一概念以来,FLPs便一直受到来自不同领域化学工作者的广泛关注。在其短短十年的发展历程中,FLPs从简单的磷/硼体系,逐步扩展到13/15族Lewis酸碱体系,近期又将视野放大到了整个元素周期表;其应用也从最初的氢气的捕捉与活化发展到不对称催化氢化、高分子聚合反应、CO2催化还原等更加广泛的领域;而FLPs在配合物中的种种应用,将过渡金属催化和主族活化有机地结合在一起,从而有助于更高效地活化各种小分子。
发展至今,FLPs体系已经在许多领域显示出独特而优越的反应性能,但正如文中分析的那样,FLPs领域还存在很大的发展空间。在实际应用层面,某些FLPs反应的条件需要进一步优化,FLPs催化剂的合成需要进一步探索,简单高效FLPs体系的大规模应用和试剂化的可行性也值得进一步探讨。而从原理层面上讲,FLPs与过渡金属的融合还有进一步的发展空间,利用FLPs的活化能力与过渡金属的多重价态构建连续反应体系将是未来研究的重点。同时,利用FLPs体系进行C―H键活化的设想也将为有机化学工作者带来新的工作思路。计算化学手段的应用也将为不对称FLPs催化体系的设计、过渡金属FLPs体系的设计、催化反应机理的研究等方面提供有力的支持。总而言之,相信经过人们进一步的摸索和完善,FLPs化学必将走向成熟。
参考文献
[1]Lewis,G.N.Valence and the Structure of Atoms and Molecules;Chemical Catalogue Company,Inc.:New York,1923.
[2]Welch,G.C.;Juan,R.R.S.;Masuda,J.D.;Stephan,D.W.Science 2006,314(5802),1124.
[3]Wang,H.D.;Fröhlich,R.;Kehr,G.;Erker,G.Chem.Commun.2008,No.45,5966.
[4]Sajid,M.;Klose,A.;Birkmann,B.;Liang,L.;Schirmer,B.;Wiegand,T.;Eckert,H.;Lough,A.J.;Fröhlich,R.;Daniliuc,C.G.;Grimme,S.;Stephan,D.W.;Kehra,G.;Erker,G.Chem.Sci.2013,4(1),213.
[5]Erős,G;Mehdi,H.;Pápai,I.;Rokob,T.A.;Király,P.;Tárkányi,G.;Soós,T.Angew.Chem.Int.Edit.2010,49(37),6559.
[6]Chase,P.A.;Jurca,T.;Stephan,D.W.Chem.Commun.2008,No.14,1701.
[7]Chen,D.;Klankermayer,J.Chem.Commun.2008,No.18,2130.
[8]Chen,D.;Wang,Y.;Klankermayer,J.Angew.Chem.Int.Edit.2010,49(49),9475.
[9]Sumerin,V.;Chernichenko,K.;Nieger,M.;Leskelä,M.;Rieger,B.;Repo,T.Adv.Synth.Catal.2011,353(11-12),2093.
[10]Stephan,D.W.;Greenberg,S.;Graham,T.W.Inorg.Chem.2011,50(4),1233.
[11]Liu,Y.;Du,H.J.Am.Chem.Soc.2013,135(18),6810.
[12]Chen,E.Y.-X.Top.Curr.Chem.2013,334,239.
[13] Murahashi,S.;Nozakura,S.I.;Hatada,K.;Takeuchi,S.;Aoki,T.Sen-iken.Nenpo.1960,13,99.
[14]Ikeda,M.;Hirano,T.;Tsuruta,T.Makromol.Chem.1971,150(1),127.
[15]Kitayama,T.;Masuda,E.;Yamaguchi,M.;Nishiura,T.;Hatada,K.Polym.J.1996,No.24,817.
[16] Zhang,Y.;Miyake,G.M.;Chen,E.Y.-X.Angew.Chem.Int.Edit.2010,49(52),10158.
[17]Zhang,Y.;Miyake,G.M.;John,M.G.;Falivene,L.;Caporaso,L.;Cavallo,L.;Chen,E.Y.-X.Dalton.Trans.2012,41(30),9119.
[18]Bolig,A.D.;Chen,E.Y.-X.J.Am.Chem.Soc.2001,123(32),7943.
[19] Ning,Y.;Zhu,H.;Chen,E.Y.-X.J.Organomet.Chem.2007,692(21),4535.
[20]Jun,K.-W.;Lee,K.-W.Ind.Eng.Chem.Res.2001,40(5),1355.
[21] Momming,C.M.;Otten,E.;Kehr,G.;Fröhlich,R.;Grimme,S.;Stephan,D.W.;Erker,G.Angew.Chem.Int.Edit.2009,48(36),6643.
[22]Zhao,X.;Stephan,D.W.Chem.Commun.2011,47(6),1833.
[23]Ashley,A.E.;Thompson,A.L.;O′Hare,D.Angew.Chem.Int.Edit.2009,48(52),9839.
[24]Sumerin,V.;Schulz,F.;Nieger,M.;Leskelä,M.;Repo,T.;Rieger,B.Angew.Chem.Int.Edit.2008,47(32),6001.
[25]Berkefeld,A.;Piers,W.E.;Parvez,M.J.Am.Chem.Soc.2010,132(31),10660.
[26] Ménard,G.;Stephan,D.W.J.Am.Chem.Soc.2010,132(6),1796.
[27]Courtemanche,M.-A.;Légaré,M.-A.;Maron,L.;Fontaine,F.G.J.Am.Chem.Soc.2014,136(30),10708.
[28]Wang,T.;Stephan,D.W.Chem.-Eur.J.2014,20(11),3036.
[29]Kuzu,I.;Krummenacher,I.;Meyer,J.;Armbruster,F.;Breher,F.Dalton.Trans.2008,No.43,5836.
[30]Harman,W.H.;Peters,J.C.J.Am.Chem.Soc.2012,134(11),5080.
[31] Forrest,S.J.K.;Clifton,J.;Fey,N.;Pringle,P.G.;Sparkes,H.A.;Wass,D.F.Angew.Chem.Int.Edit.2015,54(7),2223.
[32]Neu,R.C.;Otten,E.;Lough,A.;Stephan,D.W.Chem.Sci.2011,2(1),170.
[33]Chapman,A.M.;Haddow,M.F.;Wass,D.F.J.Am.Chem.Soc.2011,133(45),18463.
[34]Gnanaprakasam,B.;Zhang,J.;Milstein,D.Angew.Chem.Int.Edit.2010,49(8),1468.
[35]Zhang,J.;Leitus,G.;Ben-David,Y.;Milstein,D.J.Am.Chem.Soc.2005,127(31),10840.
[36]Gunanathan,C.;Ben-David,Y.;Milstein,D.Science 2007,317(5839),790.
中图分类号:O6-1;G64
doi:10.3866/PKU.DXHX20160401www.dxhx.pku.edu.cn
*通讯作者,Email:bj@pku.edu.cn
Research Progress on Frustrated Lewis Pairs Chemistry
LIU Han-Yu YANG Zhong-Tian YU Zi-Di MI Tian-Xiong BIAN Jiang*
(College of Chemistry and Molecular Engineering,Peking University,Beijing 100871,P.R.China)
Abstract:Frustrated Lewis Pairs(FLPs)represent a class of Lewis acids and bases possessing unique reactivities.Since their discovery,FLPs have obtained extensive attention and have grown rapidly on a broad range of studies.This paper highlights the application of FLPs in the fields of asymmetric hydrogenation,polymerization and catalytic reduction of CO2.Transition-metal FLPs and complexes containing FLPs ligands are also reviewed.Finally the trend in the development of FLPs is also discussed.
Key Words:Frustrated Lewis Pairs;Asymmetric hydrogenation;Ionic polymerization;Catalytic CO2reduction;Transition-metal catalytic systems
