羧基导向C―H官能团化/脱羧偶联反应研究进展
2016-07-25石先莹刘课艳
石先莹 刘课艳
(陕西师范大学化学化工学院,西安710119)
·今日化学·
羧基导向C―H官能团化/脱羧偶联反应研究进展
石先莹*刘课艳
(陕西师范大学化学化工学院,西安710119)
摘要:以邻或对位取代苯甲酸为原料,通过羧基导向的芳香羧酸邻位碳氢键官能团化继而发生脱羧反应,在原羧基的邻位引入官能团,可以合成传统付-克反应难以合成的间位取代芳香化合物。在此类反应中,羧基充当无痕导向基的功能。本文综述了基于过渡金属催化羧基无痕导向的芳香羧酸脱羧偶联策略,形成新C―C、C―杂键的研究进展。
关键词:芳香羧酸;脱羧偶联;过渡金属催化;C―H官能团化;羧基导向
过渡金属催化的交叉偶联反应能够有效地构筑C―C、C―杂键,为传统芳烃亲电取代反应难于合成的取代芳香化合物提供了一条新的途径[1-3]。其中,脱羧偶联是羧酸(盐)经金属催化脱羧后形成的活性有机金属中间体与其他底物进行的反应。20世纪60年代,Nilsson与其合作者[4,5]发现使用化学当量的Cu2O可促进邻硝基苯甲酸在沸腾的喹啉溶液中释放CO2气体,加入的碘苯可与中间体反应给出偶联产物。21世纪初,Myers等[6]首次报道了芳基羧酸和苯乙烯、丙烯酸酯的脱羧Heck偶联反应,引起了脱羧偶联反应的研究热潮。
脱羧偶联反应可以克服其他偶联反应在区域选择性上的一些局限性,且芳香酸具有价廉、易得、稳定、易存储、品种多样等诸多优点,因此,脱羧偶联反应作为构建C―C、C―杂键的重要手段受到了极大关注[7],已广泛应用于合成生物活性分子和药物分子等。例如,2006年,Goossen等[8]把分子
根据所形成新键的位置不同,可将脱羧偶联反应分为两类:(1)新键的形成选择性地发生在羧基的原位(图1,途径1);(2)羧基邻位形成新键的脱羧偶联反应(图1,途径2)。在途经2中,羧基作为导向基,在其邻位引入官能团后,继而脱去,即羧酸作为无痕导向基,充当可移去导向基的功能。通过此途径,可以由邻或对位取代苯甲酸类化合物出发制备通过传统付-克反应难以合成的间位取代芳香化合物,从而弥补了传统芳烃亲电取代反应的不足(传统芳烃亲电取代反应不能在活化的邻、对位定位基的间位直接引入基团)。
在文献报道的大多数脱羧偶联反应中,新C―C、C―杂键的形成往往选择性地发生在原羧基所在的位置。基于原位脱羧偶联策略,文献相继报道了几类具有重要用途化合物的一步高效合成新方法,主要包括:联苯[9-11]、芳基炔[12-14]、烯烃[15-18]等。文献中有关原位脱羧偶联反应的综述已有报道[19-21],本文就近年来基于过渡金属催化羧基无痕导向的脱羧偶联策略,形成新C―C、C―杂键的研究进展进行介绍。

图1 羧酸参与的脱羧偶联反应途径
1 烯基化反应
传统的芳烃付-克烃基化反应可以在活化基的邻、对位引入烃基,而不能直接引入烯基。2010年,Satoshi等[22]在铑催化剂和银盐氧化剂的存在下,以芳香羧酸和苯乙烯为原料,通过羧基导向的邻位烯基化及脱羧串联反应,经过一锅两步法得到一系列间位取代的二苯乙烯衍生物(图2)。

图2 羧基导向C―H键烯基化/脱羧反应
2008年,Satoshi等[23]利用钯催化剂,一水合醋酸铜为添加剂,实现了吲哚-3-甲酸羧基邻位的C―H烯基化/脱羧偶联反应(图3)。利用此催化体系,也可以实现吡咯-2-甲酸、噻吩-2-甲酸、呋喃-2-甲酸、苯并噻吩-2-甲酸、苯并呋喃-2-甲酸的邻位烯基化/脱羧偶联反应。随后,他们发现芳香羧酸和苯乙烯及其衍生物也可在铑催化下发生类似反应[24]。

图3 羧基邻位C―H键烯基化/脱羧反应
2014年,Quan和Xie[25]发展了以铱为催化剂,在醋酸银以及醋酸铜存在下,采用碳硼烷和炔为原料,羧基邻位的B―H键可以选择性发生烯基化及脱羧反应(图4)。通过对反应过程中关键中间体的确认,他们提出了反应可能经过铱调节的B―H键活化、炔的插入、质子化以及脱羧等串联途径。
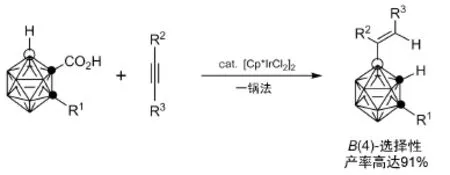
图4 羧基导向B―H键烯基化/脱羧反应
2 芳基化反应
联苯类化合物是很多天然产物、药物中间体、人造染料、高分子材料以及农药的重要骨架,因此发展简单、高效合成联苯类化合物的方法一直备受化学家们的关注。2011年,Cornella等[26]以羧酸和碘苯为原料,通过间位选择性芳基化反应,合成了一系列间位取代的联苯类化合物(图5),这种新方法为一些成本高的Suzuki偶联反应提供了一种高效的、廉价的补充。但此方法需使用邻位带有取代基的芳香羧酸,且反应中有酸性碘化氢副产物形成。
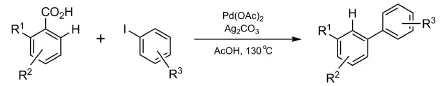
图5 羧基导向C―H键芳基化/脱羧反应
近年来,自然界丰富、廉价的C―H化合物使过渡金属催化的C―H键功能化反应为传统C―C、C―杂键的形成提供了又一极具吸引力的选择,基于C―H键功能化的偶联反应作为一种重要的合成手段得到了迅速的发展,受到了广泛的关注[27-34]。最近,苏伟平等[35]、游劲松等[36]先后报道了铑催化下,芳香羧酸和芳香杂环类化合物通过羧基邻位的C―H芳基化、脱羧偶联反应构建2-芳基杂芳环化合物的新方法(图6)。其中,2-芳基杂芳环化合物是很多天然产物、生物活性分子的重要骨架。采用这种方法,苏伟平等合成了克级规模的生物活性分子3,5-取代的2-芳基噻吩化合物。特别一提的是,克级规模的生物活性化合物通过传统的合成方法很难得到。
他们提出反应可能按照如图7所示的历程进行。反应中催化剂活性中心铑先和羧酸中的氧原子配位,促使羧基邻位C―H键活化并形成金属环化物A,环化物A和杂芳烃反应形成重要的杂芳烃-金属环化物中间体B,B经过还原消除、脱羧以及脱酸质子化反应后给出最终产物。

图6 羧基邻位交叉脱氢/脱羧反应

图7 羧基邻位芳基化/脱羧反应可能的催化机理
3 酰胺化反应
2015年,采用铑、钌催化剂,石先莹与其合作者[37,38]建立了芳香羧酸邻位酰胺化/脱羧偶联反应合成间位取代的N-芳基苯甲酰胺化合物的新方法(图8)。与传统预活化的羧酸衍生物和胺在高温下直接缩合方法相比,新方法具有简单、高效、原料廉价且来源广泛等显著优势。这一发现为难以合成的间位取代N-芳基苯甲酰胺提供了一种高效的合成新途径。

图8 羧基导向C―H键酰胺化/脱羧反应
在反应过程中,首先经历了过渡金属催化的羧基辅助邻位碳氢官能团化反应,形成的金属环化物和异氰酸酯发生Grignard-type的加成反应,加成产物脱去一分子二氧化碳,质子化后给出目标产物(图9)。

图9 羧基邻位酰胺化/脱羧反应可能的催化机理
4 串联环化反应
通过羧酸和炔的串联环化反应,可以合成多环化合物。例如,将吲哚3-甲酸、(苯并)呋喃或吡咯-2-羧酸和炔以1:2物质的量比混合[39],在Pd(OAc)2/Cu(OAC)2·H2O催化剂存在下,通过羧基导向的C―H官能团化、脱酸反应可以合成一系列的咔唑衍生物,其中一些化合物具有重要的固态荧光性能。羧酸和炔通过分子间脱氢、脱羧偶联/环化反应,可形成菲类化合物(图10)[40]。经过对机理的研究,Wang等[40]提出反应可能经历了羧基导向的C―H键活化、烯基化、脱羧、还原消除过程。

图10 羧酸和炔的串联环化反应
5 C—杂键的形成
以羧基为无痕导向基,除了可以构建新的C―C键外,也可以形成新的C―O、C―N键。Bhadra等[41]发现间位取代的芳基醚可以由邻、对位取代的芳香羧酸盐和硼酸三烷基酯反应合成,而对位取代的芳基醚可以由间位取代的芳香羧酸盐和硼酸三烷基酯反应制备(图11)。反应不仅具有产率良好、选择性优良等优点,而且避免了昂贵的Pd、Rh等催化剂的使用。
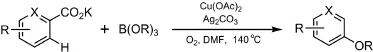
图11 羧基导向C―O新键形成/脱羧反应
Chang等[42]以邻位取代的芳香羧酸和磺酰叠氮化合物为原料,通过芳烃C―H键酰胺化、脱羧偶联反应,采取一锅两步法直接合成了间位、对位取代的N-磺酰苯胺(图12)。特别指出的是,N-磺酰苯胺类化合物通过其他的C―H官能团化反应不能直接合成。
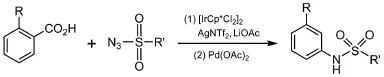
图12 羧基导向C―H键磺酰胺化/脱羧反应
6 总结与展望
羧基无痕导向的脱羧偶联反应作为构建C―C、C―杂键的重要手段,虽然已被应用于一些重要化合物的合成中,但因为反应的化学选择性问题,这类反应至今报道的比较少。此外,所用的催化体系中,价格高的过渡金属催化剂使用得较多;相反,有关廉价催化剂却鲜有报道。而且所报道的脱羧偶联反应无一例外都为均相反应,这就导致了价格昂贵的催化剂难以回收利用。随着原子经济性反应及可持续化学概念的提出,在温和条件下,选择可回收的、高效的催化剂进行普适性更高的羧基无痕导向脱羧偶联反应研究,将对化学工作者提出更高的挑战。
参考文献
[1]郭兴伟,李志平,李朝军.化学进展,2010,22(7),1344.
[2]陈垚,赖文勇,解令海,黄维.科学通报,2011,56(13),995.
[3]王乃兴.有机化学,2011,31(8),1319.
[4]Nilsson,M.Acta Chem.Scand.1966,20(2),423.
[5]Björklund,C.;Nilsson,M.Acta Chem.Scand.1968,22(8),2585.
[6]Myers,A.G.;Tanaka,D.;Mannion,M.R.J.Am.Chem.Soc.2002,124(38),11250.
[7]Rodríguez,N.;Goossen,L.J.Chem.Soc.Rev.2011,40(10),5030.
[8]Goossen,L.J.;Deng,G.;Levy,L.M.Science 2006,313(5787),662.
[9]Lange,P.P.;Goossen,L.J.;Podmore,P.;Underwood,T.;Sciammetta,N.Chem.Commun.2010,47(12),3628.
[10]Zhang,F.;Greaney,M.F.Org.Lett.2010,12(21),4745.
[11]Goossen,L.J.;Lange,P.P.;Rodríguez,N.;Linder,C.Chem.Eur.J.2010,16(13),3906.
[12]Fang,P.;Li,M.;Ge,H.J.Am.Chem.Soc.2010,132(34),11898.
[13]Li,M.;Ge,H.Org.Lett.2010,12(15),3464.
[14]Zhang,W.W.;Zhang,X.G.;Li,J.H.J.Org.Chem.2010,75(15),5259.
[15]Zhang,S.L.;Fu,Y.;Shang,R.;Guo,Q.X.;Liu,L.J.Am.Chem.Soc.2010,132(2),638.
[16]Fu,Z.J.;Huang,S.J.;Su,W.;Hong,M.Org.Lett.2010,12(21),4992.
[17]Sun,Z.M;Zhang,J.;Zhao,P.Org.Lett.2010,12(5),992.
[18]Zhou,X.;Luo,J.;Liu,J.;Peng,S.;Deng,G.J.Org.Lett.2011,13(6),1432.
[19]Shang,R.;Liu,L.Sci.China Chem.2011,51(11),1670.
[20]付拯江,李兆杰,熊起恒,蔡琥.有机化学,2015,35(5),984.
[21]戴建军,王光祖,徐小岚,许华建.有机化学,2013,33(5),2460.
[22]Satoshi,M.;Koji,H.;Tetsuya,S.;Miura,M.Org.Lett.2010,12(24),5776.
[23]Maehara,A.;Tsurugi,H.;Satoh,T.;Miura,M.Org.Lett.2008,10(6),1159.
[24]Mochida,S.;Hirano,K.;Satoh,T.;Miura,M.Org.Chem.2011,76(9),3024.
[25]Quan,Y.;Xie,Z.J.Am.Chem.Soc.2014,136(44),15513.
[26]Cornella,J.;Righi,M.;Larrosa,I.Angew.Chem.Int.Edit.2011,50(40),9429.
[27]Engle,K.M.;Mei,T.S.;Wasa,M.;Yu,J.Q.Acc.Chem.Res.2012,45(6),788.
[28]Gao,K.;Yoshikai,N.Acc.Chem.Res.2014,47(4),1208.
[29]Arockiam,P.B.;Bruneau,C.;Dixneuf,P.H.Chem.Rev.2012,112(11),5879.
[30]Arnold,P.L.;McMullon,M.W.;Rieb,J.;Kuehn,F.E.Angew.Chem.Int.Edit.2015,54(1),82.
[31]Girard,S.A.;Knauber,T.;Li,C.J.Angew.Chem.Int.Edit.2014,53(1),74.
[32]Neufeldt,S.R.;Sanford,M.S.Acc.Chem.Res.2012,45(6),936.
[33]Li,B.;Dixneuf,P.H.Chem.Soc.Rev.2013,42(13),5744.
[34]Thirunavukkarasu,V.S.;Kozhushkov,S.I.;Ackermann,L.Chem.Commun.2014,50(1),29.
[35]Zhang,Y.;Zhao,H.;Zhang,M.;Su,W.Angew.Chem.Int.Edit.2015,54(12),3817.
[36]Qin,X.;Sun,D.;You,Q.;Cheng,Y.;Lan,J.;You,J.Org.Lett.2015,17(7),1762.
[37]Shi,X.Y.;Liu,K.Y.;Fan,J.;Dong,X.F.;Wei,J.F.;Li,C.J.Chem.Eur.J.2015,21(5),1900.
[38]Shi,X.Y.;Dong,X.F.;Fan,J.;Liu,K.Y.;Wei,J.F.;Li,C.J.Sci.China Chem.2015,58(8),1286.
[39]Yamashita,M.;Hirano,K.;Satoh,T.;Miura,M.Org.Lett.2009,11(11),2337.
[40]Wang,C.;Rakshit,S.;Glorius,F.J.Am.Chem.Soc.2010,132(40),14006.
[41]Bhadra,S.;Dzik,W.I.;Gooßen,L.J.Angew.Chem.Int.Edit.2013,52(10),2959.
[42]Lee,D.;Chang,S.Chem.Eur.J.2015,21(14),5364.
中图分类号:O625.1;G64
doi:10.3866/PKU.DXHX201508003
*通讯作者,Email:shixy@snnu.edu.cn
基金资助:中央高校基本科研业务费专项资金项目(GK201503030)间的脱羧偶联应用到了Suzuki偶联反应中,高效合成了多种药物重要中间体——联苯类化合物。
Carboxyl-Directed C―H Functionalization and Subsequent Decarboxylative Coupling Reactions of Aromatic Acids
SHI Xian-Ying*LIU Ke-Yan
(School of Chemistry&Chemical Engineering,Shaanxi Normal University,Xi′an 710119,P.R.China)
Abstract:Employing ortho or papa-substituted benzoic acids as starting materials,meta-substituted aromatic compounds,which are much more difficult to be prepared by the traditional methods,can be generated via an ortho C―H functionalization directed by carboxyl and subsequent ipso decarboxylation. In these reactions,carboxyl acts as the function of traceless-directing group.This paper reviews the recent progress in the transition-metal-catalyzed decarboxylative coupling reactions of aromatic acids to construct C―C and C―heteroatom bonds with carboxyl as the traceless-directing group.
Key Words:Aromatic carboxylic acids;Decarboxylative coupling;Transition-metal-catalyzed;C―H Functionalization;Carboxyl-directing
