环戊烷-甲烷水合物生成过程的温度特性
2016-07-14胡亚飞蔡晶李小森中国科学院广州能源研究所中国科学院天然气水合物重点实验室广东省新能源和可再生能源研究开发与应用重点实验室广东广州50640中国科学院大学北京00049
胡亚飞,蔡晶,李小森(中国科学院广州能源研究所,中国科学院天然气水合物重点实验室,广东省新能源和可再生能源研究开发与应用重点实验室,广东 广州 50640;中国科学院大学,北京 00049)
研究开发
环戊烷-甲烷水合物生成过程的温度特性
胡亚飞1,2,蔡晶1,李小森1
(1中国科学院广州能源研究所,中国科学院天然气水合物重点实验室,广东省新能源和可再生能源研究开发与应用重点实验室,广东 广州 510640;2中国科学院大学,北京 100049)
摘要:研究了环戊烷-甲烷水合物生成过程中的温度变化,分析了体系的热量损失。在不同初始温度(4℃、8℃和12℃)、压力(2MPa、4MPa、6MPa、8MPa和10MPa)和进气方式(一次性进气、连续进气和间歇进气)的条件下,测定了釜内温度,对比了以上各因素对釜内最高温度(Tmax)与釜内最大温升(ΔTmax)的影响。实验表明,Tmax主要受压力和进气方式影响,初始温度对其影响不明显;ΔTmax受初始温度、压力和进气方式影响显著。在间歇进气方式下,初始温度越低、压力越高,ΔTmax越大。其中,在初始温度为4℃、压力为10MPa、进气时间间隔为30min的间歇进气方式下,ΔTmax可达16.5℃。此外,由热量分析发现,体系的主要热量损耗表现为体系向环境中的散热。因此,提高保温层的绝热性能,有利于提高水合物生成热的热量有效利用率。
关键词:水合物;甲烷;环戊烷;生成热;最大温升
第一作者:胡亚飞(1989—),男,硕士研究生。联系人:李小森,博士,研究员,博士生导师,主要从事天然气水合物方面的研究。E-mail lixs@ms.giec.ac.cn。
气体水合物是由轻烃、二氧化碳、硫化氢等气体小分子与水分子在低温或高压条件下形成的一种非化学计量的笼型包络化合物[1]。其中,主体水分子通过氢键构建出笼型孔穴结构,客体分子填充于孔穴结构之中,通过范德华力与水分子结合,从而形成结构稳定的水合物。气体水合物的形成是一种特殊的相变过程,在水合物形成过程中物质由气相或液相转移到水合物固相,并伴随着相变热的释放。根据水合物的相变特性,衍生出了一系列的水合物应用技术,如利用水合物进行海水淡化[2-3]、二氧化碳(CO2)捕获与封存[4-5]以及将水合物作为蓄冷工质应用于空调蓄冷系统中等[6-7]。其中,水合物在空调蓄冷技术中的应用是基于水合物形成和分解过程中的相变热特性。然而,由于纯气体水合物形成条件苛刻、水合物生成时间普遍较长,导致气体水合物形成过程中释放的水合物相变热热量有限且热量损耗严重。为提高水合物相变热热量的释放,通常采用添加水合物促进剂加快水合物形成速率的方式来增加水合物相变热的释放,降低单位时间内的热量损失。常见的热力学促进剂通过参与水合物结构的构建、降低形成水合物的相平衡条件来加速水合物的形成;动力学促进剂通过降低溶液的表面张力、增加气液接触面积的方式来加速水合物的形成速率,从而增加水合物相变热的释放量。相关合适的水合物热力学促进剂主要有环戊烷(CP)、四丁基溴化铵(TBAB)和四氢呋喃(THF)等[8-12],动力学促进剂有十二烷基硫酸钠(SDS)[13]和十二烷基苯磺酸钠(SDBS)[14]等。
根据相变热在气体水合物形成与分解过程中的释放特点,将水合物形成过程中释放的热量称为水合物生成热,将水合物分解过程中吸收的热量称为水合物分解热。对水合物形成分解过程中相变热的定量分析一直是水合物热特性研究的热点和难点。目前,水合物相变热数值的确定主要有两种方法,分别为:直接法——利用差示扫描量热仪(DSC)实验测定水合物分解热[15-17];间接法——利用Clausius-Clapeyron方程计算获得水合物分解热[18-19]。其中,直接法为实验方法,借助DSC实验设备测定相应条件下水合物形成或分解过程中的热流变化,从而获得对应水合物的理论相变热数值;而间接法为理论计算方法,在水合物相平衡数据的基础上,根据Clausius-Clapeyron方程计算得到理论相变热数值∆H。然而,以上两种确定水合物相变热的方法都只从水合物热特性参数的基础研究出发,而非出于水合物相变热热量实际应用价值的研究目的,尤其针对保温系统中,因水合物生成热释放而引起的体系温度特性研究较少。中国科学院天然气水合物重点实验室李小森课题组[20-21]首次提出了一种直接利用水合物生成热加热海水形成热盐水进行天然气水合物原位开采的方法,结果表明该方法相对传统的注热盐水开采法具有明显的优越性,拓展了气体水合物相变热的应用领域。其中水合物形成过程中生成热所引起的体系温度变化是影响该水合物原位开采方法开采效率的重要因素之一。
因此,本文以釜内温度变化为考察对象,针对环戊烷-甲烷水合物的形成体系,分别考察了不同初始温度、压力以及进气方式对环戊烷-甲烷水合物生成过程中釜内温度变化的影响,进而分析其对釜内最高温度以及釜内最大温升的影响,并对水合物生成体系的热量损失进行了详细的分析。
1 实验部分
1.1 实验材料
实验所用气体为摩尔分数 99.9%的纯甲烷气体,由佛山华特气体有限公司提供;环戊烷纯度为99.0%,由成都贝斯特试剂有限公司提供;实验用水为电导率18.25mΩ/cm的去离子水,由南京超纯水技术有限公司提供的超纯水设备制备。
1.2 实验装置
图1为水合物生成过程中温度测定的实验装置示意图,该装置主要由进排液模块、供气模块、反应釜、低温室以及数据采集模块五部分组成。反应釜由 316不锈钢制成,釜高为 100mm,釜内径为40mm,釜内总体积为125mL,最大承压为15MPa。如图1所示,釜内自上往下分布有3个Pt100热电偶,其测量值分别为T1、T2、T3,温度的测量精度为±0.01℃。反应釜顶部安置有型号为trafag8251的压力传感器,用以测量反应釜内压力,测压范围为0~40MPa,测量精度为±0.02MPa。反应釜釜体用热导率较小的气凝胶保温材料[λ<0.02W/(m·K),25℃时]进行包裹,气凝胶层的包裹厚度为50mm。供气模块中高压贮气罐总体积为 8L,最大承压为45MPa,顶部安置有Pt100热电偶和型号为setra5310的压力传感器,测压范围为 0~40MPa,测量精度为±0.02MPa。低温室温度由氟利昂吊顶式冷风机进行控制调节,其温度通过Pt100热电偶监控,记录为Te。其中,实验中的所有压力和温度值由计算机软件自动采集并记录。

图1 实验装置示意图
1.3 实验方法
环戊烷-甲烷水合物是结构 II型水合物[22],理想比例构成的环戊烷-甲烷水合物的分子表达式为8CP·16CH4·136H2O。因此,实验采用环戊烷与水的摩尔比例为1∶17,按反应液体积为70mL转换为体积,则 CP与 H2O的体积分别为 16.5mL和53.5mL,本文所有实验均在此比例下进行,去离子水与环戊烷的混合反应液体积均为70mL。
水合物生成实验开始前,调节低温室温度为实验设定值,实验中需要使用的气体与液体均放置于低温室中,保证实验前得到充分冷却。实验开始前,用增压泵将贮气罐中甲烷气增压至30MPa。使用真空泵对反应釜抽真空,采用负压注入去离子水清洗反应釜3次。随后对反应釜再次抽真空,将环戊烷与水的混合反应液注入反应釜,静置 10min。之后从釜底鼓泡通入甲烷气体至实验设定压力,测定并记录釜内温度变化。实验中分别考察了初始温度为4℃、8℃和12℃,压力为2MPa、4MPa、6MPa、8 MPa和10MPa,以及进气方式分别为一次性进气、连续进气和间歇进气对釜内温度的影响。实验过程中,储气罐中出口处甲烷气的压力通过调节调压阀维持恒定。当储气罐内的压力保持30min不变化时,则认为该条件下的水合物生成过程结束。水合物生成实验结束后,打开釜顶控制阀排空釜内自由气,并将反应釜放置于20℃的条件下分解水合物,利用排水集气法确定水合物分解相中甲烷气的体积。最后,排出釜内反应液,清洗反应釜并将反应釜移至低温室,调节低温室温度为实验设定温度,重复进行下一组实验。
2 结果与讨论
2.1 初始温度的影响
在压力为10MPa、进气方式为连续进气的实验条件下,讨论了不同初始温度T0(4℃、8℃和12℃)对釜内温度T1、T2、T3的影响,确定不同初始温度条件下的釜内最高温度和釜内最大温升。在水合物生成过程中将测定的反应釜内最高温度称为釜内最高温度(Tmax),釜内最高温度与初始温度之差为釜内最大温升(ΔTmax)。Te为低温室温度,在±1.5℃的温度范围内波动。
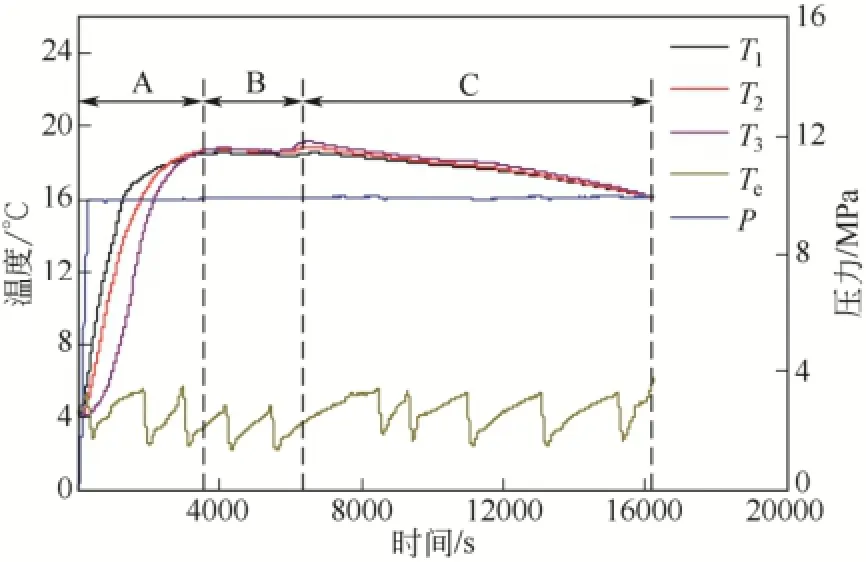
图2 初始温度为4℃、压力为10 MPa条件下釜内温度T1、T2、T3的变化图
图2为初始温度为4℃、压力为10 MPa时,水合物生成过程中釜内温度和压力随水合物生成时间的变化图。如图2所示,在水合物生成过程中,釜内温度出现了 3种不同的变化趋势:在 0~60min内,釜内温度快速上升,如A段所示;在60~105min内,釜内温度维持稳定,如B段所示;105~270min内,釜内温度不断下降,如C段所示。如图2所示,A段中釜内不同位置的温度T1、T2、T3均由4℃快速上升至18.8℃附近。由此可见,随着甲烷气体从反应釜底部不断地鼓入,釜压从0增至实验压力10MPa,环戊烷-甲烷水合物大量生成,在生成过程中伴随着大量生成热的释放。同时,由于本实验使用的反应釜(如图1所示)包裹有保温层,使得水合物生成过程中释放的相变热累积在反应体系中,致使釜内温度不断升高,从而实现加热整个反应体系的目的。在 A段水合物生成过程中,T1、T2、T3瞬时速率均呈现出先增加后降低的变化趋势。这是由于大量生成的水合物不断地在气液界面堆积,减小了水合物形成过程中的气液接触面积,与此同时,受釜内温度上升的影响,新水合物形成驱动力降低,水合物生成速率随之减慢。然而,不断有气体鼓入釜内,气体从反应釜底部上升到气液界面,此过程中的气体扰动作用对气液接触面的更新具有一定的积极作用。在以上3个抗衡因素的作用下,T1、T2、T3瞬时速率会不断发生变化。A段前期,水合物在气液界面还未形成明显的堆积,此时气液接触面积大且气体扰动致使气液接触面更新较快,因而釜内温度瞬时速率大;随着水合物在气液界面上不断地堆积,水合物的生成因气液接触面的更新受限和釜内温度升高的双重影响,从而使得釜内温度瞬时速率不断降低,直至B段,釜内温度瞬时速率为零。
此外,对比图2中A段T1、T2、T3各自的温度变化发现,相同时间点釜内温度呈现出T1> T2> T3的变化特点。由图1可知,T1温度传感器位于气液界面附近,T2、T3温度传感器均位于溶液中。综合图1与图2可知,环戊烷-甲烷水合物最初在气液界面处生成,随着反应的不断进行,水合物生成界面不断地向溶液中移动。因此,水合物生成过程中释放的大量相变热首先加热 T1,随后依次加热 T2和T3。在釜压维持恒定压力15min后,T1为15.5℃,T3为 8.0℃,此时 T1与 T3间的温差值最大,高达7.5℃。A段后期,釜内温度T1、T2、T3逐渐接近,最后均达到了18.8℃。此时,体系中气液接触面更新受限、水合物生成驱动力减小,环戊烷-甲烷水合物生成速率减慢。当单位时间内水合物生成热释放热量与反应釜损失热量相近时,釜内温度T1、T2、T3开始稳定,从而转为B段的釜内温度稳定阶段。B段中,由于底部鼓入的气体与水合物堆积层中间隙水之间存在着一定的有效气液接触面,因此,此阶段中仍有少量水合物生成。然而,受釜内温度高和反应釜热量损失的影响,釜内温度T1、T2、T3变化不明显。随着反应釜内水合物在堆积层中不断生成,水合物浆液越来越致密,气液接触越来越困难,使得水合物生成过程中的传质阻力不断增大,从而导致水合物生成速率减慢甚至停止。水合物生成热释放热量小于反应釜向低温环境散失的热量,釜内温度T1、T2、T3由稳定的B阶段进入下降的C段。图3为压力为10MPa,初始温度分别为4℃、8℃、12℃时,以釜内温度T2为例详述水合物生成过程中釜内温度随时间的变化图。从图3可知,在初始温度为8℃和12℃条件下,釜内温度T2的温度变化趋势与图2所示的釜内温度T1、T2、T3的变化趋势相同,都经历了图2所示的A、B、C三段不同的温度变化过程。然而,在初始温度为12℃的条件下,釜内温度上升段(A段)持续的时间长达165 min,远大于初始温度为 4℃和 8℃条件下 60min和64min。并且在初始温度为12℃条件下的A段前期,釜内温度变化较小且呈现出明显的温度平台,而在A段后期,釜内温度变化明显且温升速率快。由图3可知,在初始温度为12℃条件下气体初始通入反应釜便出现第一段温升(0~15min),该温升较小,釜内温度达 13.4℃后便由温升转为温降,经过了100min的缓慢温降后,釜内温度降至13.1℃;随后出现第二段温升,第二段温升(105~165min)结束时釜内温度升至20.6℃。引起第一段温升的热量主要来源为甲烷气在反应液中的溶解热,而引起第二段温升的热量来源为大量环戊烷-甲烷水合物生成所释放的相变热。在初始温度为12℃时,水合物的形成驱动力下降,形成水合物需要较长的诱导时间,因此在A段前期出现了一段较长时间的温度平台,且釜内温升仅为1.1℃。在4℃和8℃初始条件下,因初始温度降低,体系中形成水合物的温度驱动力增大,水合物形成的诱导时间缩短,A段釜内温度 T2呈现快速上升的变化趋势。此外,从图 3可知,在初始温度为12℃条件下Tmax达20.6℃,高于初始温度为4℃和8℃条件下的Tmax达18.8℃和20.0℃。这可能是因为初始温度提高后,对应的低温环境温度较高,在生成水合物的过程中体系向低温环境中散失的热量减少所致。

图3 压力为10 MPa,初始温度分别为4℃、8℃、12℃条件下釜内温度T2的对比图
根据相平衡曲线可知,在10MPa压力条件下的环戊烷-甲烷水合物体系中,理想的釜内最高温度Tmax值为该相平衡点的温度,约为30℃[23]。然而,在实际的实验过程中,受釜内温度升高、水合物生成速率减慢和体系热量损失的影响,致使反应体系中Tmax存在温度上限,该上限值小于30℃,同时反应体系中存在ΔTmax。图4为实验压力为10MPa的恒压条件下,初始温度不同所引起的ΔTmax以及甲烷气体消耗量的对比图。从图4可知,相同压力条件下初始温度越高,ΔTmax越小,ΔTmax值由初始温度为4℃条件下的14.8℃降低到初始温度为12℃条件下的 8.6℃。即降低反应初始温度,有利于提高ΔTmax。如图4所示,初始温度为4℃、8℃和12℃条件下,甲烷气体的消耗量分别为5.66L、5.24L和4.60L,即初始温度越高,甲烷气体消耗量越小。甲烷气体的消耗量越小,说明环戊烷-甲烷水合物的生成量越小,水合物生成过程中释放的相变热热量越小,影响着ΔTmax数值的大小。
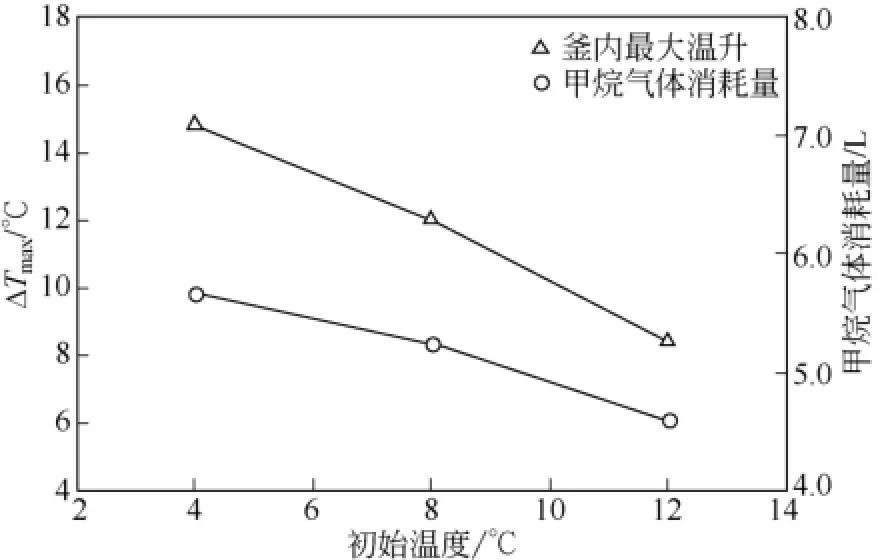
图4 初始温度分别为4℃、8℃、12℃条件下的釜内最大温升ΔTmax和甲烷气体消耗量对比图
2.2 压力的影响
在初始温度为 4℃、进气方式为连续进气的条件下,讨论了不同压力(2MPa、4MPa、6MPa、8MPa 和10MPa)对釜内温度T1、T2、T3的影响,对比不同压力条件下的Tmax和ΔTmax值。图5为反应压力分别为2MPa、4MPa、6MPa、8MPa和10MPa以及连续进气条件下的釜内温度变化图。从图5可知,不同压力条件下,各体系的温升时间起点以及釜内温度达到 Tmax所需时间不同,呈现出低压(2MPa 和4MPa)和高压(6MPa、8MPa和10MPa)两种不同的变化特点。在2MPa和4MPa的低压条件下,从反应开始到釜内温度出现明显变化至少需要17h,水合物形成诱导时间长,且Tmax分别仅为5.6℃和12.6℃;而在6MPa、8MPa和10MPa的较高压力条件下,水合物形成诱导短,均在4h之内就完成了如图2所示的A段釜内温度快速上升阶段,且Tmax分别达到了 16.8℃、17.8℃和 18.8℃。由此可见,压力对釜内温度的影响显著。相同初始温度条件下,压力越高,水合物的形成驱动力越大,水合物生成速率越快,水合物的生成量越大,越能有效地提高体系的Tmax值。由图5可知,低压条件下反应釜内温度的升高呈现了3个不同的变化阶段,分别表现为:①0~15min,温度变化速率快但变化幅度较小,热量来源于大量甲烷气溶解于反应液中释放的溶解热;②1~17h,温度变化速率缓慢且变化幅度不大,热量来源于形成少量水合物时所释放的水合物生成热;③17~25h,温度变化速率快且变化幅度较大,热量来源于釜内快速形成大量水合物时释放的大量生成热。从图5中较高压力条件下釜内温度变化曲线可知,反应釜内温度的升高仅呈现了类似于图2中A段的温升变化。在6MPa、8MPa 和 10MPa压力条件下对庆的温升时间分别分为235min、96min和60min,此时Tmax维持稳定的时间(即对应图2中B段的时间)分别为241min、47min 和45min。由图5可知,6MPa压力条件下Tmax维持稳定的时间最长。这是因为6MPa压力下生成的水合物致密程度较低,水合物堆积层中有一定量间隙水存在,在压力稳定的情况下,间隙水逐渐转化为水合物,持续释放出少量相变热,同时,与8MPa和10MPa压力条件相比,6MPa压力下的Tmax与低温环境间的温差较小,釜内热量向低温环境中散失的速率低。
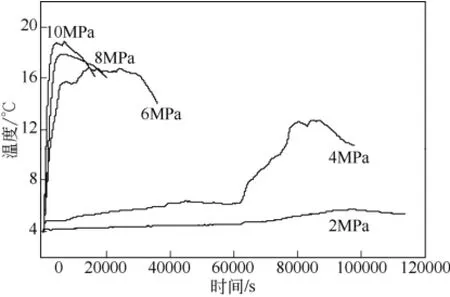
图5 初始温度为4℃、不同压力条件下釜内温度T2的变化对比图
从图6可知,随着压力的升高,ΔTmax呈现递增的趋势,但增加的幅度随着压力的升高而降低。在4MPa、8MPa与10MPa 3种压力条件下,甲烷气体消耗量相近,但ΔTmax差异明显。这是因为甲烷气体消耗量相近时,压力越高,生成水合物的压力驱动力越大,水合物生成速率越快,Tmax越高,ΔTmax也越大。在6MPa压力条件下,环戊烷-甲烷水合物的甲烷气体消耗量为7.04L,大于8MPa和10MPa压力条件下甲烷气体消耗量5.36L和5.66L。这是因为在6MPa压力下,当釜内温度达Tmax时,因水合物致密程度低,此时有大量间隙水转化到环戊烷-甲烷水合物中,生成了更多的环戊烷-甲烷水合物。由图6可知,6MPa、8MPa与10MPa压力条件下的ΔTmax分别为12.8℃、13.8℃和14.8℃,即压力越高,ΔTmax越大。由此可见,压力、水合物生成量以及散热速率都影响着釜内温升的大小,其中压力对环戊烷-甲烷水合物生成过程中釜内温升的影响最为显著。

图6 初始温度为4℃、不同压力条件下的釜内最大温升ΔTmax和甲烷气体消耗量对比图
2.3 进气方式的影响
在初始温度为4℃、压力为10MPa,讨论了不同进气方式对釜内温度T1、T2、T3的影响,确定不同进气方式条件下的Tmax和ΔTmax。对于不同进气方式实验,进气方式有一次性进气、连续进气和间歇进气3种。其中,一次性进气方式为直接增压至实验压力后停止进气;连续进气方式为直接增压至实验压力后维持实验压力恒定的持续进气;间歇进气方式为分段增压进气至实验压力,其中,分段压力采用2MPa、4MPa、6MPa、8MPa、10 MPa,本实验进气间隔采用10min和30min两种。
2.3.1 一次性进气方式与连续进气方式
图7为一次性进气下体系的釜内温度和压力变化图。根据釜内压力的变化过程,可将釜内压力划分为釜压急剧上升段(a段)、釜压下降段(b段)、釜压快速下降段(c段)和釜压缓慢下降段(d段)。由图7可知,釜内温度在b段几乎不变化,在c段时,因水合物大量生成,釜内热量集中,釜内温度快速增加至12.1℃。釜内温度达12.1℃后,釜内压力下降明显变慢(d段),但釜内温度仍在缓慢升高,此时釜压下降的原因主要为釜内水合物仍在缓慢生成而不断消耗甲烷气体;釜温升高至最高温度12.5℃后,釜内温度由温升转为温降,此时釜压缓慢下降的主要为:因釜内温度下降,由气体状态方程可知釜压也会同步下降。此外,与图2所示的连续进气方式中釜内温度能快速达到最大值不同,图7中釜内温度随时间的变化呈现升高、稳定、再升高、降低的变化趋势。可将一次性进气下体系的釜内温度升高分为两段:通入甲烷气后便出现第一次温升(升高约3.0℃);70min之后出现第二次温升(升高约5.5℃)。实验中测得一次性进气体系中形成水合物所消耗的甲烷气体量为 2.50L,是约为连续进气体系中甲烷气体消耗量的一半,说明一次性进气条件下形成的水合物量较少。对比图2和图7可知,在初始温度和实验压力相同的条件下,连续进气方式比一次性进气方式更有利于提高体系的Tmax和ΔTmax值。
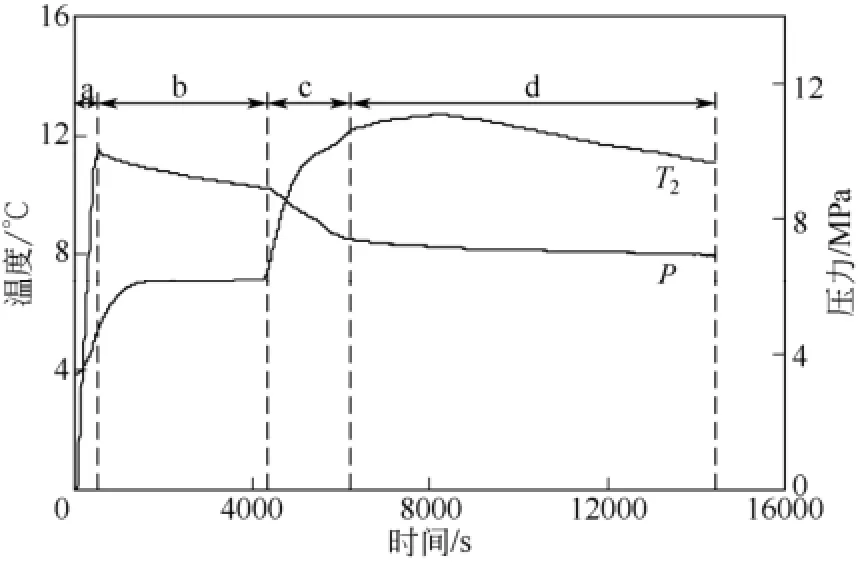
图7 初始温度为4℃,一次性进气下体系的釜内温度和压力变化图
2.3.2 间歇进气方式
图8与图9分别给出了间隔时间为10min和30min的间歇进气方式下釜内温度和压力变化图。在图8与图9中,如S段所示,釜压未达实验压力10MPa前,釜压按照梯状方式增加,釜内温度也呈现了类似的增加趋势。当釜压达到10MPa后,釜内温度急剧上升且迅速达到各自的 Tmax值 20.4℃和20.5℃。这是因为在釜压达 10MPa时,釜内温度约为6.5℃,而6.5℃对应的环戊烷-甲烷水合物相平衡压力略小于 0.16MPa[19,23],此时釜内压力驱动力较大,并且S段中有5次较大的增压扰动,两个因素均有利于釜内水合物的快速生成,因此,釜内温度上升快速且幅度较大。然而,随着釜内温度不断升高,当釜温达Tmax后,反应釜内压力驱动力减小,水合物生成速率降低,水合物生成过程中释放的相变热热量有限,同时受体系向低温环境散热的影响,釜内温度不再升高并呈现出下降的趋势。
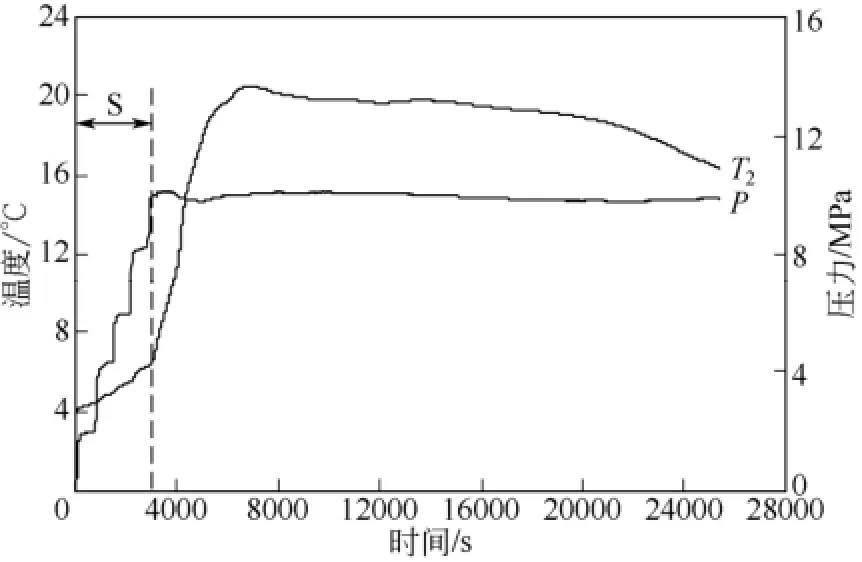
图8 初始温度为4℃、间隔时间为10min的间歇进气下体系的釜内温度和压力变化图

图9 初始温度为4℃、间隔时间为30min的间歇进气下体系的釜内温度和压力变化图

表1 不同间隔时间间歇进气方式下的实验数据表
表1为不同间隔时间间歇进气方式下的实验数据表,其中,定义开始注气到釜内温度达最大值所用的时间为表观温升时间t1,Δti为间歇进气的间隔时间。对比图2、图8与图9可知,t1依次增加,主要与间隔时间Δti有关。将 t1与总间隔时间 4Δti之差定义为实际温升时间t2,由表1可知t2数值相近。t3为反应釜内Tmax维持稳定的时间,由表1可知t3随间隔时间增大而增加。与表1对应的不同间隔时间的间歇进气方式下釜内温度变化曲线如图10所示。由图10和表1可知,间隔时间为0min、 10min和30min下的Tmax数值分别为18.8℃、20.4℃和 20.5℃,对应的 t3则分别为45min、116min和143min。可见间歇进气方式比连续进气方式下的Tmax与t3值均要大。间歇进气方式中间隔时间的长短对Tmax数值的影响较小,但对t3的大小影响显著。较长的间隔时间对应着更大的t3值,t3值越大意味着Tmax维持的时间越长,即釜内维持高温段的时间越长,这种釜内温度变化特性有利于水合物生成热热量的有效利用。同时,由表1中可知,间歇进气方式比连续进气方式消耗的甲烷气体量 Vm更大,在达到实验压力前加入适当的压力扰动有利于增加水合物的总生成量。综合可知,间歇进气方式能达到比连续进气方式更大的 Tmax值和ΔTmax值,并且较大的间隔时间能较大程度地提高Tmax维持稳定的时间t3。因此,间歇进气方式为一种较优的进气方式。
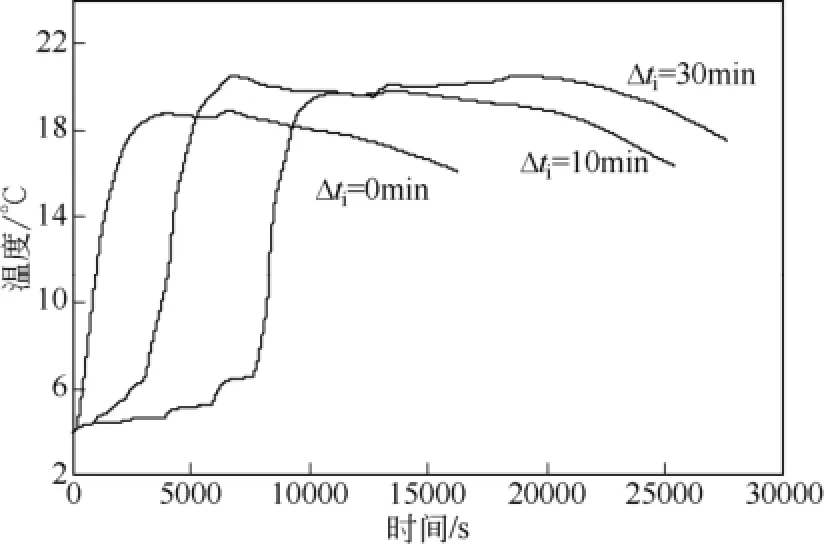
图10 初始温度为4℃,不同间隔时间间歇进气下体系的釜内温度变化图
3 热量分析
本实验开始前,气体、液体及反应装置均处于控制温度为T0条件下的低温室中。因反应釜保温层的保温性能有限,体系在整个实验过程中存在一定的热量损耗。其中,体系热量的损耗包括体系热量向钢釜层和保温层的转移以及向低温环境的散失。在实验过程中,释放的水合物生成热部分用来加热釜内水合物浆与自由甲烷气,其余的热量转移到钢釜层和保温层以及低温环境中。反应持续的时间越长,体系向周围低温环境散失的热量就越多。因此,釜内最高温度 Tmax和釜内最大温升ΔTmax受水合生成速率和体系散热速率两方面的影响。
釜内环戊烷-甲烷水合物生成热的热量去向具体表现在以下5个方面:①釜内反应液和水合物因温度升高而吸收的热量Qh,w,c;②釜内自由甲烷气因温升而吸收的热量Qm;③316不锈钢釜温度升高吸收的热量Qs;④保温层温度升高吸收的热量Qa;⑤体系向低温环境散失的热量Qd。另外,通入釜内的甲烷气溶解于反应液中会释放一定量的溶解热,但由于该溶解热远小于反应过程中釜内释放的水合物生成热,以下计算忽略溶解热部分,并假设5个方面的热量全部来自于反应釜内水合物生成热的总热量Qf,即有Qf=Qh,w,c+Qm+Qs+Qa+Qd。并将有效热量Qh,w,c与Qf的比值定义为热量有效利用率,其表达式如式(1)。
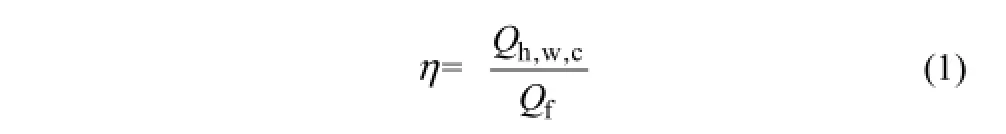
以图2条件下水合物的生成过程为例,定义体系温降较明显处为实验反应结束点,详细分析该过程中释放的水合物生成热热量在整个装置中的具体分布情况。该条件下,生成的水合物所消耗的甲烷气的体积为 5.66L,由状态方程 PV = ZnRT得 n = 0.236mol,其中T为室温20℃。假设本实验中最终生成的水合物全部为环戊烷-甲烷水合物,并且分子构成全部为理想分子式8CP·16CH4·136H2·O,则水合物晶体结构中含有的环戊烷与水的总量分别为0.118mol和2.006mol。
环戊烷-甲烷水合物释放的总生成热热量如式(2)。

纯水体系中环戊烷-甲烷水合物生成热在温度范围286~292K间变化较小,此时qf为129~130 kJ/mol[18-19],此处取为 129.5kJ/mol,则由式(1)得Qf=30.56kJ。
对于Qh,w,c的计算,公式为式(3)。

式中,cp,h、cp,w和cp,c分别环戊烷-甲烷水合物、水和环戊烷的定压比热容,近似取cp,h=2.8J/(g·K)[1,26]、cp,w=4.2J/(g·K)、cp,c=1.8J/(g·K);mh、mw和mc分别环戊烷-甲烷水合物、水和环戊烷的质量,计算得mh=48.1g、mw=17.4g、mc=4.0g;Δt为釜内反应液和水合物的最大温升值,Δt=14.8℃。
对于Qm的计算,公式为式(4)。
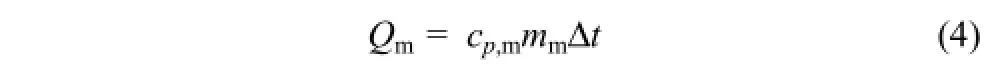
式中,cp,m为甲烷气体的定压比热容,取cp,m= 3.1J/(g·K);mm为甲烷气体的质量,计算得 mm= 4.27g;Δt为釜中甲烷气的最大温升值,Δt=14.8℃。
对于Qs的计算,公式为式(5)。
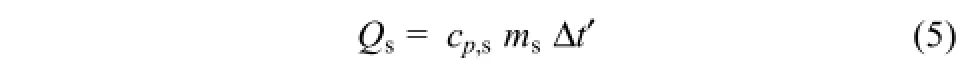
式中,cp,s为316不锈钢定压比热容,取cp,s为0.50J/(g·K);ms为 316不锈钢的质量,计算得 ms=520.2g;Δt′为钢釜的平均温升,因钢釜热导率和热扩散系数均较大,钢釜厚度薄,侧壁厚2.5mm,底部厚10mm,热阻Rs小,紧邻钢釜层外包裹有保温层,体系向低温环境散热热流量Φ 也较小,钢层ΔT=RsΦ 较小,可认为钢釜整体处于均一温度,此处Δt′近似取值为比Δt低0.8℃,即Δt′=14.0℃。
对于Qa的计算,公式为式(6)。
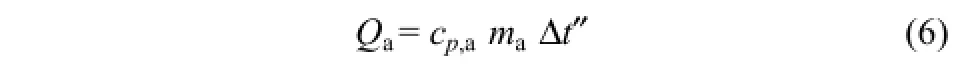
式中,cp,a为气凝胶定压比热容,取 cp,a=0.56 J/(g·K)[27];ma为气凝胶的质量,计算得ma=662.5g;Δt′为保温层的平均温升,对于保温层,散热热流量Φ 虽小,但热阻Rs较大,因保温层包裹圆柱型反应釜,为空心圆柱型结构,其温度分布复杂,将保温层的平均温度采用等效温度简化计算,此处Δt″近似取值为比Δt′低6℃,即Δt″=8.0℃。
对于Qd的计算,因直接计算体系向低温环境的散热量较为复杂,此处采用间接计算方式,如式(7)。
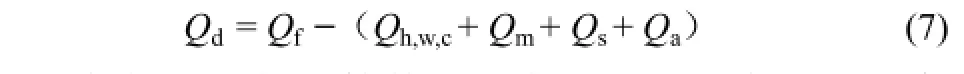
各热量具体计算数值见表2。表2中列出了实验过程中水合物生成热在不同初始温度(4℃、8℃和12℃)下的各热量具体数值和各热量占水合物生成热总热量的百分比及各热量有效利用率。
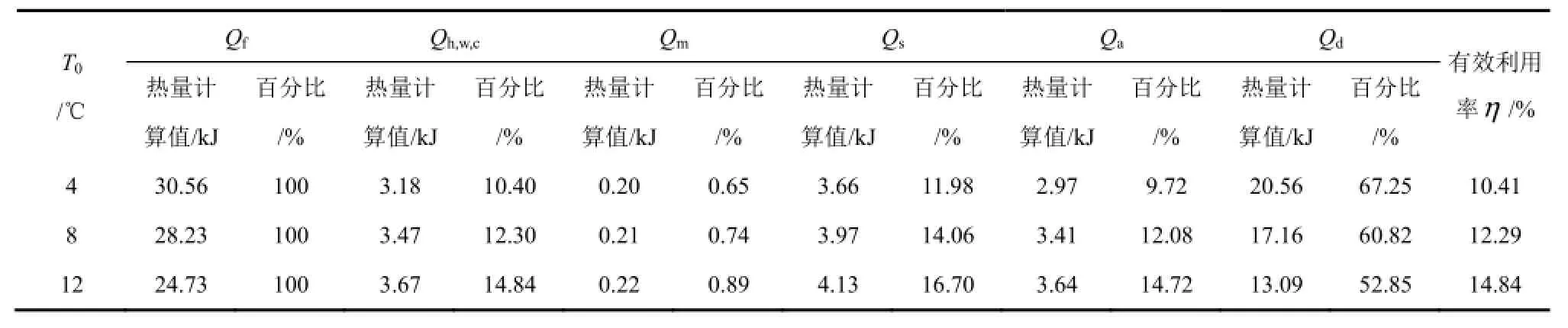
表2 反应体系各热量计算数值及百分比
由表2可知,3种初始温度下的热量有效利用率η在10.41%~14.84%之间,说明水合物生成热总热量的有效利用率有限。仅有少于15%的热量用于釜内液体的加热,而绝大部分热量(占总热量的比例高达85%以上)被反应装置自身吸收和釜体向低温环境扩散。其中,釜体向环境扩散的热量占52.85%~67.25%,为生成热热量损失的最主要去向。由于实验前体系整体与低温环境温度相同,此时钢釜层与保温层在实验过程中吸收的热量无法避免。从表2可知,在不同初始温度下,Qh,w,c、Qs、Qa三者的值相近,但明显小于Qd。初始温度增加,Qd占总热量的比例减小,而 Qh,w,c、Qs、Qa的比例在增大,热量有效利用率η也增大,可见提高初始温度可减少体系热量向低温环境的扩散。由以上热量分析可知,提高反应釜的绝热性能,能有效地增大水合物生成热的热量有效利用率,并使 Tmax和ΔTmax均有一定程度的增加。
4 结 论
本文进行了环戊烷-甲烷水合物生成过程温度特性的实验研究,考察了初始温度、压力和进气方式对釜内温度的影响,并对体系中水合物生成热的热量去向进行了分析,得出了以下几点结论。
(1)初始温度越低,Tmax越低,ΔTmax越高。
(2)压力越高,Tmax越高,ΔTmax也越高。
(3)相对于连续进气和一次性进气方式,间歇进气方式可达到更大的Tmax和ΔTmax。
(4) 在初始温度为4℃、压力为10MPa和间断进气时间为 30min的反应条件下,ΔTmax可达16.5℃。
(5)实验过程中,较小一部分热量留在反应釜内,绝大部分热量会向低温环境扩散,提高装置的绝热性能可使Tmax和ΔTmax均有一定程度的增加。
符 号 说 明
cp——定压比热容,J/(g·K)
H——气体水合物的相变热,kJ/mol
m——质量,g
n——物质的量,mol
Qh,w,c——釜内反应液和水合物因温度升高而吸收的热量,kJ
Qm——釜内自由甲烷气因温升而吸收的热量,kJ
Qs——316不锈钢釜温度升高吸收的热量,kJ
Qa——保温层温度升高吸收的热量,kJ
Qd——体系向低温环境散失的热量,kJ
Qf——水合物生成热的总热量,kJ
qf——单位物质的量的水合物生成热热量,kJ
R——通用气体常数,8.314 J/(mol·K)
Ti——釜内温度(i=1,2,3),℃
T0——反应初始温度,℃
Te——低温室温度,℃
Tmax——釜内最高温度,℃
ΔTmax——釜内最大温升,℃
t1——表观温升时间,min
t2——实际温升时间,min
t3——反应釜内釜内最高温度维持稳定的时间,min
Δt——釜内温升,℃
Δti——间歇进气的间隔时间,min
Δt′——316不锈钢釜层的温升,℃
Δt″——气凝胶层的温升,℃
Vm——甲烷气体的消耗量,L
Z——压缩因子
λ——热导率,W/(m·K)
η——热量有效利用率
下角标
a—— 环戊烷-甲烷水合物
c——水
h——环戊烷
m——甲烷
s——316不锈钢
w——气凝胶
参 考 文 献
[1] SLOAN E D,KOH C A. Clathrate hydrates of natural gases[M]. Florida,US:CRC Press,2007.
[2] 任宏波,相凤奎,张磊,等. 水合物法海水淡化技术应用进展[J]海洋地质前沿,2011(6):74-78.
[3] GHALAVAND Y,HATAMIPOUR M S,RAHIMI A. A review on energy consumption of desalination processes[J]. Desalination and
[4] LINGA P,ADEYEMO A,ENGLEZOS P. Medium-pressure Water Treatment,2015,54(6):1526-1541. clathrate hydrate/membrane hybrid process for postcombustion capture of carbon dioxide[J]. Environmental Science &
[5] LI X S,XU C G,CHEN Z Y,et al. Hydrate-based pre-combustion Technology,2007,42(1):315-320. carbon dioxide capture process in the system with tetra-n-butyl
[6] BI Y H,GUO T M, ZHU T G,et al. Influence of volumetric-flow ammonium bromide solution in the presence of cyclopentane[J]. Energy,2011,36(3):1394-1403. rate in the crystallizer on the gas-hydrate cool-storage process in a new gas-hydrate cool-storage system[J]. Applied Energy,2004,78
[7] 焦丽君,孙志高,赵之贵,等. 添加剂对水合物蓄冷过程影响探讨[J]. 科学技术与工程,2014,14(32):217-220.
[8] AMAN Z M,OLCOTT K,PFEIFFER K,et al. surfactant adsorption and interfacial tension investigations on cyclopentane hydrate[J]. Langmuir,2013,29(8):2676-2682. (1):111-121.
[9] LV Q N,LI X S,XU C G,et al. Experimental investigation of the formation of cyclopentane-methane hydrate in a novel and large-size bubble column reactor[J]. Industrial & Engineering Chemistry Research,2012,51(17):5967-5975.
[10] OYAMA H,SHIMADA W,EBINUMA T,et al. Phase diagram,latent heat,and specific heat of TBAB semiclathrate hydrate crystals[J]. Fluid Phase Equilibria,2005,234(1):131-135.
[11] XU C G,ZHANG S H,CAI J,et al. CO2(carbon dioxide) separation from CO2-H2(hydrogen) gas mixtures by gas hydrates in TBAB (tetra-n-butyl ammonium bromide) solution and Raman spectroscopic analysis[J]. Energy,2013,59:719-725.
[12] STROBEL T A,TAYLOR C J,HESTER K C,et al. Molecular hydrogen storage in binary THF-H2clathrate hydrates[J]. The Journal of Physical Chemistry B,2006,110(34):17121-17125.
[13] LI X S,CAI J,CHEN Z Y,et al. Hydrate-based methane separation from the drainage coal-bed methane with tetrahydrofuran solution in the presence of sodium dodecyl sulfate[J]. Energy & Fuels,2012,
[14] SUN Z G,WANG R Z,MA R S,et al. Natural gas storage in 26(2):1144-1151. hydrates with the presence of promoters[J]. Energy Conversion and
[15] 梁德青,郭开华,樊栓狮,等. HCFC-141b气体水合物融解热的DSC测试[J]. 工程热物理学报,2002(s1):47-49. Management,2003,44(17):2733-2742.
[16] ZHANG Y,DEBENEDETTI P G,PRUD'HOMM R K,et al. Differential scanning calorimetry studies of clathrate hydrate
[17] GUPTA A,LACHANCE J,SLOAN E D,et al.Measurements of formation[J].The Journal of Physical Chemistry B,2004,108(43): 16717-16722. methane hydrate heat of dissociation using high pressure differential scanning calorimetry[J]. Chemical Engineering Science,2008,63 (24):5848-5853.
[18] CHEN Z Y,LI Q P,YAN Z Y,et al.Phase equilibrium and dissociation enthalpies for cyclopentane+ methane hydrates in NaCl aqueous solutions [J]. The Journal of Chemical & Engineering Data,
[19] LV Q N,LI X S,CHEN Z Y,et al. Phase equilibrium and 2010,55(10):4444-4449. dissociation enthalpies for hydrates of various water-insoluble organic promoters with methane[J]. Journal of Chemical & Engineering Data,2013,58(11):3249-3253.
[20] 陈朝阳,李小森,颜克凤,等. 一种开采天然气水合物的方法及装置:101016841A[P]. 2007-02-13.
[21] CHEN Z Y,FENG J C,LI X S,et al. Preparation of warm brine in 2014,53(36):14142-14157.
[22] 闫忠元,陈朝阳,李小森,等. 盐水体系中环戊烷-甲烷水合物相平衡测定与模拟[J]. 过程工程学报,2010(3):476-481. situ seafloor based on the hydrate process for marine gas hydrate thermal stimulation[J]. Industrial & Engineering Chemistry Research,
[23] SUN Z G,FAN S S,GUO K H,et al.Gas hydrate phase equilibrium data of cyclohexane and cyclopentane[J]. Journal of Chemical & Engineering Data, 2002,47(2):313-315.
[24] 孙长宇,黄强,陈光进. 气体水合物形成的热力学与动力学研究进展[J]. 化工学报,2006,67(5):1031-1039.
[25] FAN S S,LIANG D Q,GUO K H.Hydrate equilibrium conditions for cyclopentane and a quaternary cyclopentane-rich mixture[J]. Journal of Chemical & Engineering Data,2001,46(4):930-932.
[26] TOMBARI E,PRESTO S,SALVETTI G,et al. Heat capacity of tetrahydrofuran clathrate hydrate and of its components,and the clathrate formation from supercooled melt[J]. The Journal of Chemical physics,2006,124(15):154507.
[27] 周祥发,冯坚,肖汉宁,等. 二氧化硅气凝胶隔热复合材料的性能及其瞬态传热模拟[J]. 国防科技大学学报,2009,31(2): 36-40,69.
System temperature properties in the process of the cyclopentane-methane binary hydrates formation
HU Yafei1,2,CAI Jing1,LI Xiaosen1
(1Guangdong Key Laboratory of New and Renewable Energy Resenrch and Development,Key Laboratory of Gas Hydrate,CAS,Guangzhou Institute of Energy Conversion,Chinese Academy of Sciences,Guangzhou 510640,Guangdong,China;2University of Chinese Academy of Sciences,Beijing 100049,China)
Abstract:In this paper,the changes of the system temperatures and heat loss were investigated during the formation of cyclopentane-methane binary hydrates. The system temperature measurements were carried out under the conditions of initial temperatures of 4℃,8℃,and12℃,pressures of 2MPa,4MPa,6MPa,8MPa and 10MPa,and different gas injection modes (single,continuous and intermittent). The maximum temperature (Tmax) and the maximum temperature increase (ΔTmax) in the system were compared. The experimental results illustrate that the pressures and gas injection modes have significant influence on Tmaxwhile the initial temperatures,pressures and gas injection modes all significantly effect ΔTmax. Thus,the conditions of lower initial temperature,higher pressure and injecting intermittently help to increase ΔTmax. Under the condition of 4℃ and 10MPa,intermittent injection with the interval time of 30minute,the maximum value of ΔTmaxis 16.5℃. In addition,the heat analysis results indicate that the main heat loss is from the inner reactor to the outside cold environment. Therefore,improving the insulation properties of insulating layer is helpful to enhancethe heating efficiency in the process of the cyclopentane-methane binary hydrates formation.
Key words:hydrate;methane;cyclopentane;formation heat;maximum temperature increase degree
中图分类号:TQ 026
文献标志码:A
文章编号:1000-6613(2016)05-1418-10
DOI:10.16085/j.issn.1000-6613.2016.05.022
收稿日期:2015-09-28;修改稿日期:2015-11-02。
基金项目:国家杰出青年科学基金(51225603)、国家自然科学基金(51376184)、中石油-中科院高端战略联盟计划(2015A-4813-2)及中海油研究总院委托项目(CRI2015RCPS0053OCN)。
