mTOR信号通路在左旋多巴诱发异动症中的作用及机制研究
2016-07-05缪茂军陈小武曹学兵陈志斌
缪茂军 陈小武 曹学兵 陈志斌
mTOR信号通路在左旋多巴诱发异动症中的作用及机制研究
缪茂军陈小武曹学兵陈志斌
570102海口,海南医学院附属医院神经内科[缪茂军陈志斌(通信作者)陈小武];华中科技大学同济医学院附属协和医院神经内科(曹学兵)
【摘要】目的探讨mTOR(Mammalian target of rapamycin,哺乳动物雷帕霉素靶蛋白)信号通路的激活在左旋多巴诱发异动症(L-Dopa Induced Dyskinesia,LID)大鼠纹状体中的作用及机制。方法普通级SD雄性大鼠共54只,随机分为5组,8只正常组(N组),剩下8只帕金森病(Parkinson's disease)模型;异动症组10只、余均分为雷帕霉素(Rapamycin)作为药物治疗组即干预组和Rapamycin溶剂的对照组(C组)各14只;记录下对照组与干预组的行为学并进行异动症AIM评分,每周至少两次;用western blot技术测定N组、PD组、LID、C组以及R组纹状体组织的免疫印迹,验证所用NMDA受体亚基NR1(Ser896)、NR2A(Y1325)、NR2B(Y1472)位点磷酸化及PSD-95、p-PSD95(S295)磷酸化位点的抗体特异性的变化。结果(1)对照组第1,3,5天时间点行为学AIM评分与干预组无差异(P>0.05),第8天时间点行为学AIM评分与对照组比较,干预组AIM评分有下降(P<0.01),之后第9、10、13、16、18、21天2组仍然有明显差异,干预组AIM评分无反弹上升现象。(2)LID组大鼠纹状体PSD-95及p-PSD95(s295)表达水平较其他组高;干预后两者的表达水平明显下降(P<0.05)。PSD-95及p-PSD95(S295)在正常组表达水平最低,在异动症组和对照组最高,无差异(P>0.05);与正常组外其他组对比,干预组两者的表达水平明显下降(P<0.05);干预组与正常组的表达水平无明显差异(P>0.05)。(3)LID组、对照组NMDA受体亚基NR1(Ser896)、NR2A(Y1325)、NR2B(Y1472)位点磷酸化的表达水平较干预组有所增高(P均<0.05)。结论(1)mTOR参与了LID的发病,应用mTOR特异性抑制剂雷帕霉素对LID的治疗有效。(2)大鼠纹状体区的mTOR信号调理与LID的发生密切相关,mTOR激活对纹状体突触水平具有调节作用,可能通过依赖突触分子PSD-95以及NMDAR各亚型的磷酸化水平导致突触可塑性发生适应改变,最终产生和维持皮质纹状体突触“病理性LTP”,从而介导了LID发生。
【关键词】异动症帕金森病mTOR雷帕霉素纹状体突触可塑性
【DOI】10.3969/j.issn.1007-0478.2016.03.011
PD是中老年人常见的神经系统退行性运动障碍疾病,目前治疗PD的主要手段就是多巴胺(dopamine,DA)替代疗法,但长期服用后可能导致严重的并发症,其中较常见又是最难处理的一种并发症就是LID,一旦出现该症状就会长期保持,严重影响左旋多巴(L-DOPA)对PD的进一步治疗,LID被认为是最普遍的限制PD治疗疗效的副作用[1],临床上处理尚无找到有效的办法。LID的发生主要是由一些受体导致纹状体的输出的变化是从而造成纹状体多巴胺受体(dopamine receptor,DR)波动性刺激并变化引起的,但确切的机制尚不完全清楚。近年研究发现:在LID模型中,直接通路(兴奋性)MSNs内哺乳动物mTOR(Mammalian target of rapamycin,哺乳动物雷帕霉素靶蛋白)信号通路能够被持续性的激活;然而阻断 mTOR的信号通路转导不影响L-DOPA对PD的治疗作用,但是却能够明显减轻LID[2-4]。
mTOR调节的蛋白质的合成在突触可塑性调节中发挥重要作用[5]。研究发现[6],利用不同频率刺激发现皮质纹状体谷氨酸能纤维能诱发长期增益效应(LTP),皮质纹状体通路异常在LID中起重要作用,即纹状体神经元通过“病理性”LTP对异常运动的记忆,是LID产生的重要机制。在调节突触可塑性中,皮质纹状体突触可塑性的异常可能是LID发生的重要机制。因此,关键分子PSD-95及骨架分子NMDA受体功能增强参与突触可塑性,在“病理性LTP”产生中起重要作用[7],但是mTOR在“病理性LTP”中的调控涉及到PSD-95、NMDA受体这一机制目前尚不清楚。
以上提示了mTOR在蛋白合成和LID中发挥了关键作用,突触异常是LID发生的重要基础,mTOR信号通路的激活在LID中发生,并可能成为治疗LID的新靶标。因此,我们进一步猜想:作为与突触可塑性相关的整合分子如PSD-95、NMDA受体。在LID中研究,mTOR信号通路的激活,导致与LTP产生和维持有关的蛋白质合成增加如PSD-95、NMDA各种亚型NR1、NR2A、NR2B的蛋白分子磷酸化表达等,mTOR通过调节蛋白质翻译机制使突触可塑性发生适应改变,最终产生和维持皮质纹状体突触“病理性LTP”,从而介导了LID发生,而这一系列的整合分子的表达是否受mTOR信号通路的调控呢?
本研究通过脑立体定向仪注射6羟基多巴(6-hydroxydopamine,6-OHDA)制作性能稳定的PD模型,对PD模型外源性输入腹腔注射左旋多巴+苄丝肼制作LID模型,并且用mTOR特异性抑制剂-雷帕霉素干预,深入阐明mTOR信号通路在LID模型中发生的相关的行为学变化、突触可塑性相关分子作用以及突触形态学观察,从而为行为学以及突触可塑性分子水平奠定理论基础。因此,本实验通过验证mTOR在LID模型中的作用,观察mTOR信号通路的激活对LID大鼠行为学及PSD-95、S295、NMDAR亚基磷酸化水平的影响及调控,进一步探讨LID的病理机制。
1材料与方法
1.1实验动物
饲养成年普通级Sprague-Daewley rats,雄性,体重从200~250 g范围,由华中科技大学同济医学院实验动物中心提供,遵照伦理学,所有饲养符合华中科技大学同济医学院实验动物中心标准和相关管理条例。
1.2实验设计分组
设计分为5组:(1)正常观察组(Normal,N组),未做任何处理;(2)帕金森模型组(Parkinson,PD组):6-OHDA(2 ug/ul,注射8 ug)脑立体注射制作,经阿扑吗啡检测制作成功的帕金森病大鼠便纳入该组,后未做任何处理;(3)异动症组(LID组):经阿扑吗啡(为0.05 mg/kg)检测后选入成功的PD纳入,L-DOPA +Benserazide (L-DOPA 25 mg+Benserazide 6.25 mg/kg,)腹腔注射给药,连续21 d;(4)Rapamycin(剂量0.35 mg/kg用药)为干预组(Rapamycin,R组):Rapamycin干预后L-DOPA给药;(5)Rapamycin的对照组(C组):Rapamycin的溶剂(5% DMSO+5%聚乙二醇400+5%聚氧乙烯山梨糖醇酐单月桂酸酯溶液(Tween-20))作为对照,L-DOPA给药不变。
1.3药物及主要试剂
6-羟多巴(6-OHDA)、阿朴吗啡、左旋多巴甲酯、盐酸苄丝肼、雷帕霉素(rapamycin)、等均购于美国sigma公司。Anti-phospho -NR1 (Ser896) 购于 millipore公司;Anti-NMDAR2A(phospho Y1325)、Anti-NMDAR2B(phospho Y1472)购于Abcam;Anti-PSD95 (phospho S295)、Anti-PSD95 购于Epitomics;Anti-β-actin 购于santa。
1.4主要实验器材
脑立体定位仪购自深圳市瑞沃德生命科技有限公司,超声匀浆器、低温离心机、电泳仪、-70 ℃超低温冰箱等均由华中科技大学同济医学院神经系统重大疾病国家重点实验室基础医学院9楼实验室提供。
1.5偏侧帕金森病大鼠模型的制备
以右侧内侧前脑束(MFB)为目标靶区,脑立体定向注射6-OHDA制作偏侧帕金森病大鼠模型[8,9]。
1.6检测PD模型
术后2周,使用阿扑吗啡(0.05 mg/Kg),旋转>7转/min 为成功的成功PD大鼠模型[10]。
1.7LID大鼠模型的制作(L-DOPA腹腔注射)
注射阿扑吗啡3 d后,LID组大鼠给予溶液L-DOPA 25 mg+Benserazide 6.25 mg/kg于腹腔注射,1次/d,连续用药7 d,评定等达到AIM[11]的标准。
Q:作为较早进入内地的香港企业,金杯印刷见证了改革开放的高速发展,您认为改革开放为贵公司提供了怎样的发展环境?
1.8LID随机分组
LID组:继续每天L-DOPA给药;R组大鼠每天在给予L-DOPA前1h给予Rapamycin;C组给予同等剂量的安慰剂(内含5% DMSO+5%聚乙二醇400+5%聚氧乙烯山梨糖醇酐单月桂酸酯溶液(Tween-20)),同时,如LID组所述,给予含L-DOPA+Benserazide溶液同剂量腹腔注射。
1.9异动症(AIM)评分
AIM评分并录取视频交有课题负责人审校,评定标准按Monville等[11]标准, AIM目的评分分为4个部分:口面部-即下颌运动和嘴部舌头伸出;上肢-即对侧前肢重复无目的的动作;轴性-即颈部和对侧上体的扭曲姿势;运动-即与对侧转向运动)进行评定,数据顺序亦按此记录,每部分的评分等级根据行为学的有无及不自主运动的严重程度来划分,共5个等级(0~4):0分为无出现;1分为偶尔出现,少于50%的时间,即少于观察期的一半;2分经常出现,超过50%的时间,即一半以上的观察期;3分为继续存在,刺激可以停止;4分为持续存在,刺激不可使之停止。从理论上评论每只大鼠,1只大鼠1次用药后每个评分时间点最大得分为16分,120 min内最大评分为64分。记录数据以供进一步分析。
1.10Western-Blot检测纹状体PSD95、S295等表达:
在药物干预的最后1 d准备冻存管,液氮等,异动症组、干预组及对照组在给予L-DOPA药物2 h后处理,快速断头,放入冰上操作,小心解剖出右侧纹状体,称重后记录下数据并置入EP管,液氮速冻,存于-80 ℃,正常组以及PD组的纹状体标本采集同上。根据各组大鼠的总重量来配置RIPA裂解液的量,加入10倍的RIPA裂解工作液体积,研磨彻底,用超声波破碎仪匀浆3 s停1 min,提取蛋白,用BCA法测定蛋白水平,根据样品数量BCA试剂按体积比A∶B=50∶1,加入5x蛋白上样缓冲液的量为按EP中蛋白样品的体积1/4,充分混合,震荡义震荡5 min后,电磁炉设置100 ℃,待达到100 ℃时候,放入沸水中加热10 min,冷却,将样本经10%的SDS-PAGE电泳分离胶进行分离后,将胶移入PVDF膜上,右上角缺失及标记处为正面,合起,恒流300 mA 2 h将蛋白转移至PVDF膜上,转膜结束,条带放入在TBST中用摇床洗5 min,然后放入预先配置好5%的脱脂奶粉中,摇床1 h即封闭结束。稀释一抗,开始一抗孵育4 ℃过夜,一抗孵育结束。TBST洗涤3*10 min或者5*6 min。稀释二抗(用TBST稀释,不回收),开始孵育二抗,室温下约1 h,二抗孵育结束。用TBST洗涤5*6 min。在暗室中按1∶1体积吸入ECL发光液A和B试剂,于EP管中充分混匀,将PVDF膜放入暗匣中曝光,扫描曝光,保存图像,若果效果不好,用TBST洗3*5 min,用一抗二抗洗脱液洗5 min,再次封闭,重新孵育一抗过夜,继续前面步骤。
1.11统计学处理

2结果
2.1行为学评分
如图1示,随机选8只对照组与8只干预组行为学评分,在第8 d之前两组无明显差异即第1,3,5 d时间点两组间无差异(P>0.05),根据实验设计,第7 d药物干预,经雷帕霉素干预后第二天即第8 d时间点即出现显著性差异(P<0.01),后AIM评分持续下降未出现明显反弹现像(表1)。
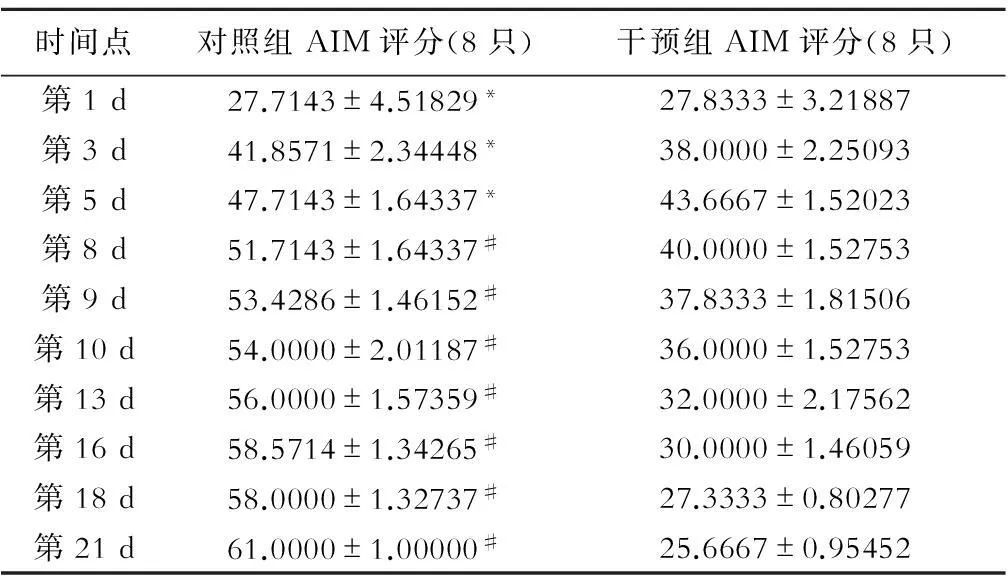
表1 2组行为学评分
注:与干预组比较,*P>0.05,#P<0.01
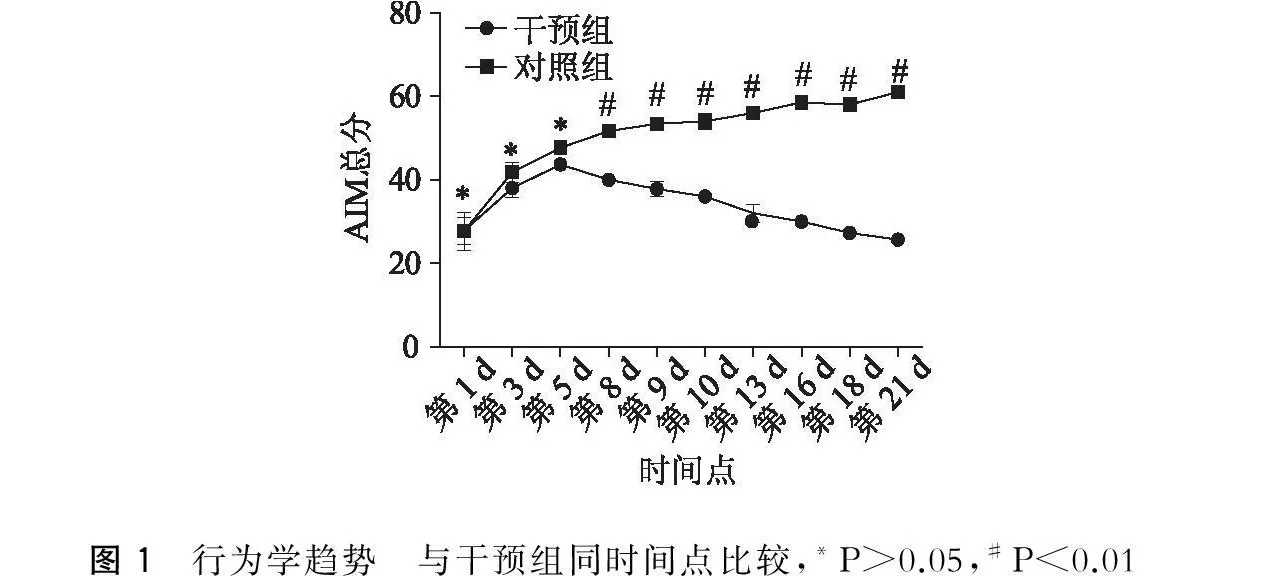
图1 行为学趋势 与干预组同时间点比较,*P>0.05,#P<0.01
2.2突触可塑性骨架分子水平Western-Blot检测
2.2.1突触可塑性骨架分子p-PSD95(s295)水平的变化LID组大鼠纹状体s295表达较正常组外的其他组高(P<0.05);N组表达水平最低;LID组和C组(P>0.05)最高,两者无明显差异(P>0.05);R组与C比较有差异(P<0.05);R与N组比较无明显差异(P>0.05)(图2)。

图2 Westernblot检测p-PSD95(s295)水平的变化 与同正常组比较,△P=0.011<0.05;同PD组比较,*P=0.176>0.05;与异动症组比较,**P=0.904>0.05;与对照组比较,***P=0.013<0.05;与干预组比较,※P=0.075>0.05
2.2.2突触骨架分子PSD-95水平的变化
PD组PSD-95表达水平与N组较高(P<0.05);LID组较PD组表达较高(P<0.05);LID组和C组(P>0.05)表达最高,两者无明显差异(P>0.05);R组与C比较有差异(P<0.05);R组与N组比较无明显差异(P>0.05)(图3)。
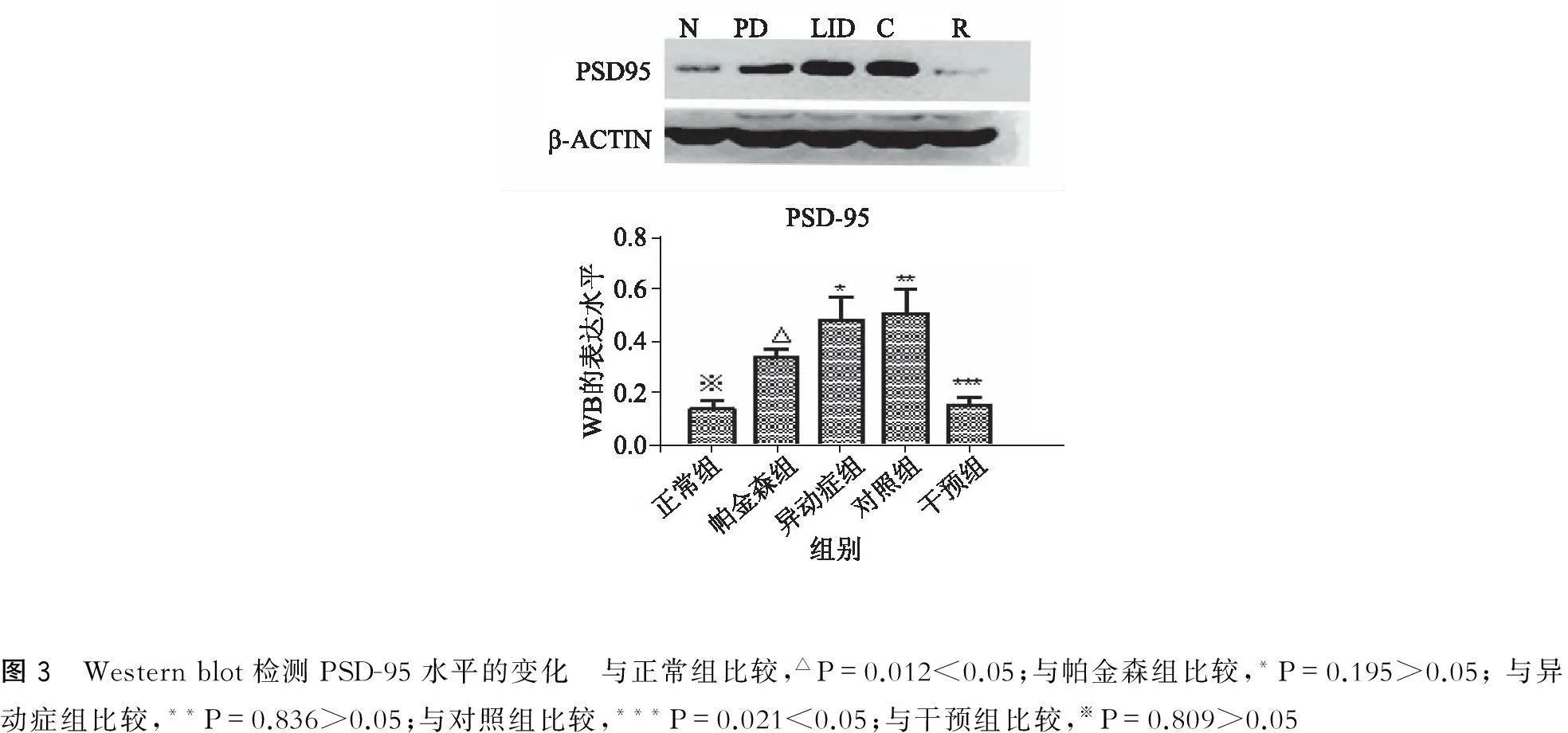
图3 Westernblot检测PSD-95水平的变化 与正常组比较,△P=0.012<0.05;与帕金森组比较,*P=0.195>0.05;与异动症组比较,**P=0.836>0.05;与对照组比较,***P=0.021<0.05;与干预组比较,※P=0.809>0.05
2.3突触可塑性关键分子水平Western-Blot检测
2.3.1mTOR信号通路激活导致的NMDA亚基NR1(ser896)磷酸化水平的变化PD组ser896磷酸化水平较N组高(P<0.01);LID组较PD组高(P<0.05);LID组和C组最高,无明显明显(P>0.05);R组与C组差异明显(P<0.01)(图4)。
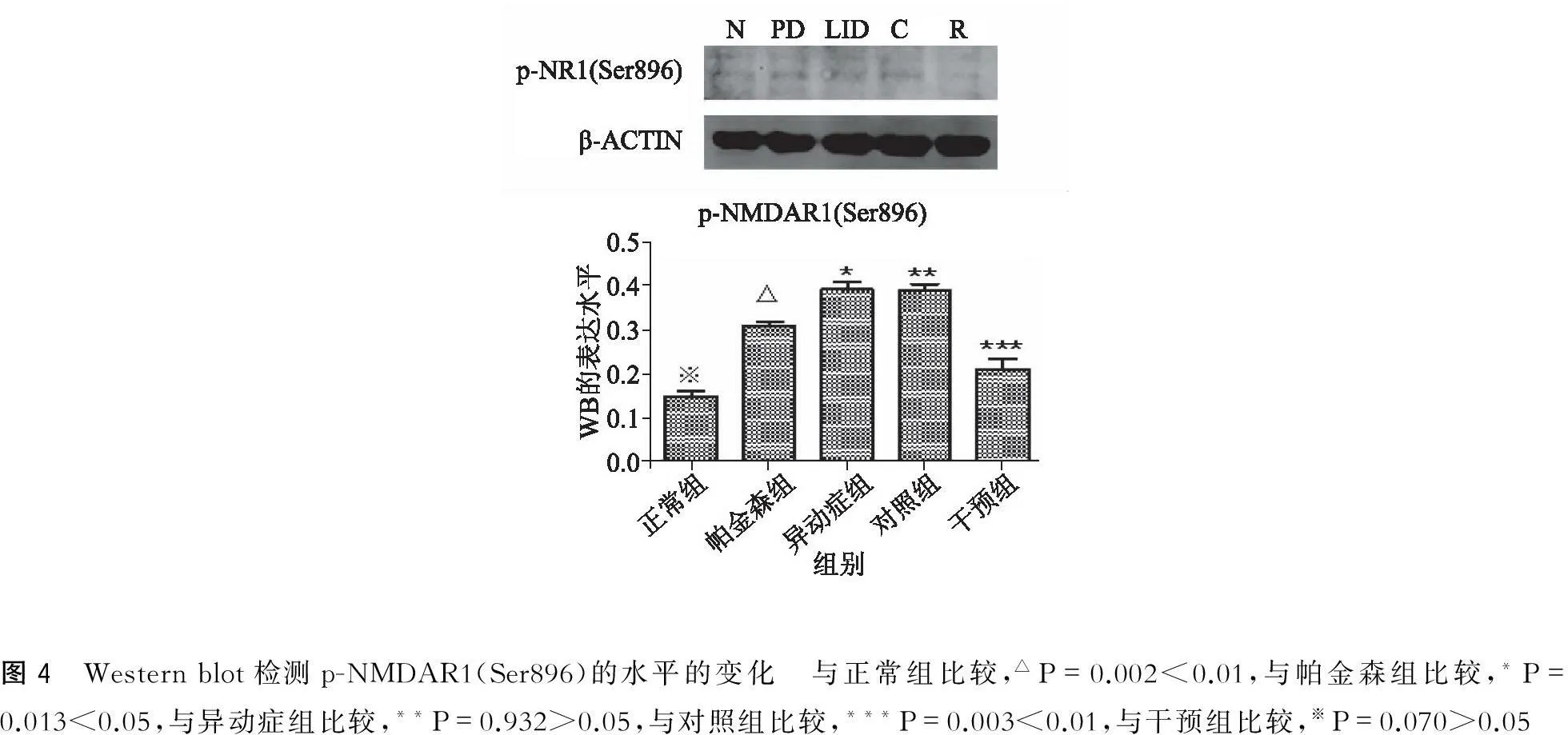
图4 Westernblot检测p-NMDAR1(Ser896)的水平的变化 与正常组比较,△P=0.002<0.01,与帕金森组比较,*P=0.013<0.05,与异动症组比较,**P=0.932>0.05,与对照组比较,***P=0.003<0.01,与干预组比较,※P=0.070>0.05
2.3.2mTOR信号通路激活导致的NMDA亚基NR2A(Y1325)磷酸化水平的变化
PD组NR2A(Y1325)磷酸化水平与N组比较,两者有明显差异(P<0.05);LID组与PD组比较,有明显差异(P<0.05);C组与LID组明显无差异(P>0.05);R组与C组比较有明显差异(P<0.01);R组比N组有明显差异(P<0.01)(图5)。
2.3.3mTOR信号通路激活导致的NMDA亚基NR2B(Y1472)磷酸化水平的变化
如图6示,PD组NR2B(Y1472)磷酸化水平较正常组高(P<0.01);LID组与PD组比较有差异(P<0.01);LID组和C组无差异(P>0.05);R组与C组比较明显差异(P<0.01);R组比N组比较有明显差异(P<0.05)。
3讨论
3.1mTOR与突触后致密区骨架分子PSD-95及S295与LID发病机制
PSD-95是在突触后密度树突棘(PSD)的骨架分子,其中,PSD-95经典角色是谷氨酸受体蛋白稳定性的主要支架部分[12]。PSD-95作为重要的支架蛋白在兴奋性突触的PSD大量富集,PSD-95中分特定的PDZ结构域大量介导了细胞内信号传导分子和细胞表面粘附分子,离子通道和受体包括NMDA受体的相互作用[13,14]。PSD-95在突触可塑性中被认可具有广泛功能,包括膜蛋白和蛋白质运输的定位调节作用。许多信号传输通过它们的胞质域。直接在突触上与PSD-95互动,形成了丰富的突触后骨架蛋白,起到了调节突触可塑性的功能。突触定位和NMDA受体的活性是由MAGUK家族蛋白(PSD-95,PSD-93,SAP-102和SAP-97)的成员调制,El-Husseini等[15]研究发现PSD-95的过度表达可以带动谷氨酸突触的成熟,它的表达可以增强谷氨酸受体的突触聚集和活动,PSD-95突触后的表现也增强了突触前终端的成熟,PSD-95的表达也增加树突棘的数量和大小,这些结果表明PSD-95的可编排突触发展,提示了PSD-95在突触稳定和可塑性的重要角色。许多病理过程是用一种或多种突触后神经递质受体的故障相关联,在PSD-95和NMDA受体的耦合与DA受体彼此间功能及调控功能中发挥重要作用。有研究显示在纹状体神经元破坏D1R和PSD-95的相互作用会降低大鼠和猕猴模型的LID发展和严重程度[16]。突触蛋白,包括受体被转运、锚定,并通过PDZ结构域的蛋白质,其中最为明显的特点是PSD-95聚集。PSD-95的提高可以改变突触可塑性的可能性,它们可能是造成帕金森病和LID原因[12]。
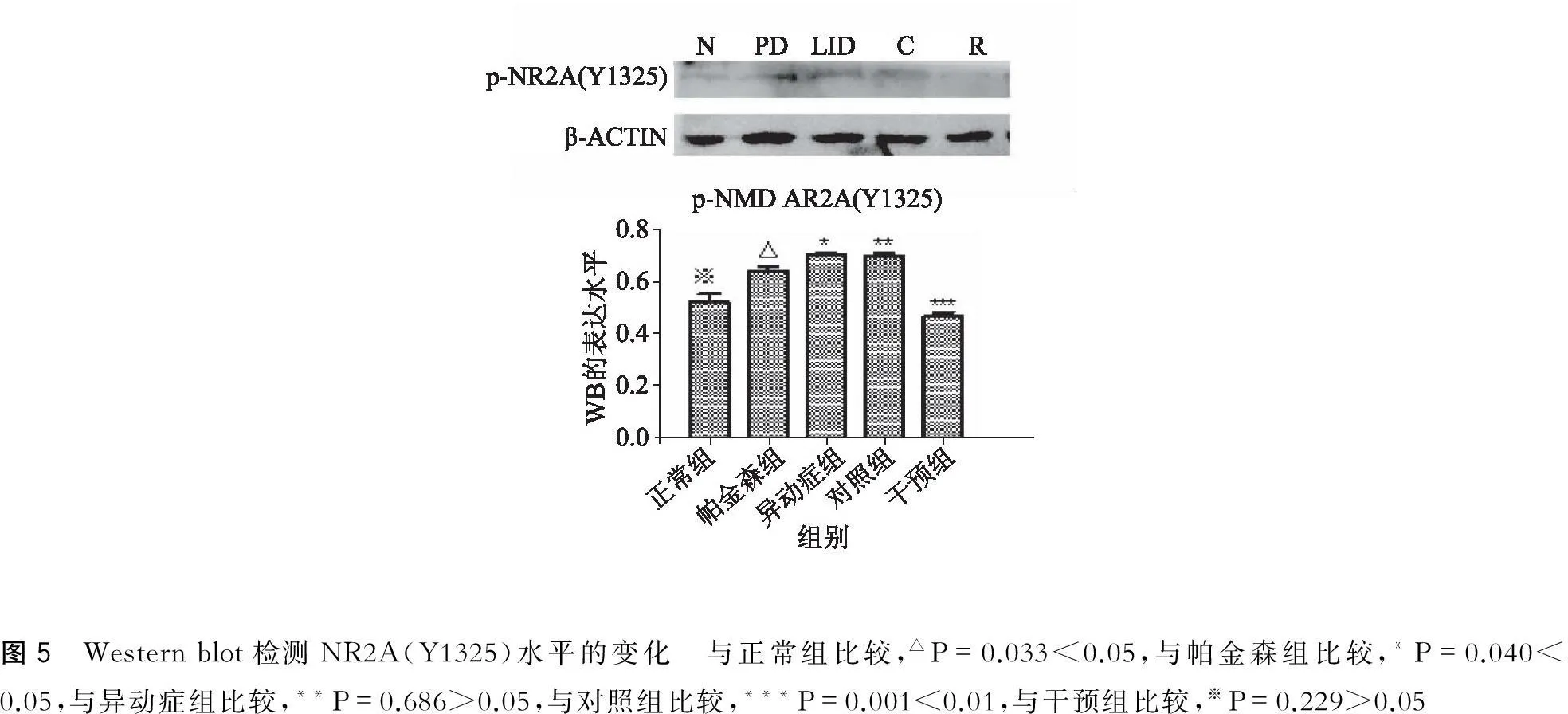
图5 Westernblot检测NR2A(Y1325)水平的变化 与正常组比较,△P=0.033<0.05,与帕金森组比较,*P=0.040<0.05,与异动症组比较,**P=0.686>0.05,与对照组比较,***P=0.001<0.01,与干预组比较,※P=0.229>0.05

图6 Westernblot检测NR2B(Y1472)水平的变化 与正常组比较,△P=0.006<0.01,与帕金森组比较,*P=0.010<0.05,与异动症组比较,**P=0.666>0.05,与对照组比较,***P=0.001<0.01,与干预组比较,※P=0.071>0.05
PSD-95的S295的磷酸化可以通过慢性活动操作来调节,S295位点的磷酸化水平反映了PSD-95的突触强度,抑制S295的磷酸化会引起响应于NMDAR处理引起的LTD,S295的磷酸化促进PSD-95和影响增强突触的积累,而LTP的诱导化学(化学LTP)的刺激增加了S295的磷酸化[17]。PSD-95是突触强度和可塑性的重要调节器,PSD-95的过度表达在培养的神经元突触增加AMPA受体集群和电流[15]。PSD-95的过表达增加了微型兴奋性突触后电流(mEPSCs)的频率,影响LTD以及LTP和关闭AMPA受体突触传递的驱动[18,19]。
本研究中发现,外源性的输入L-DOPA对6-OHDA制作的PD模型中,mTOR信号通路的激活影响到突触骨架分子PSD-95的表达,并且产生明显变化(One-Way ANOVA,P<0.05),且骨架分子PDS-95与mTOR信号通路的激活成一致性;作为骨架分子的PSD-95发生变化,PSD-95以D1和NMDA受体三元蛋白复合物的形式,并整合增强两种受体之间的的相互作用[12],最新研究表明,抑制PSD-95蛋白表达能够下调NMDA受体过度活化,从而提供了有利于LID的疗法[20]。因此本研究验证了LID中阻断mTOR信号通路会导致PSD-95及其磷酸化的水平下降,而两者都在突触可塑性具有重要的作用,这可能是mTOR信号调控底物的磷酸化水平改变了PSD-95、S295的变化。这提示了激活mTOR信号通路的途径可能依赖PSD-95以及S295的突触活动引起LID,但是两者之间具体如何相互整合突触活动尚需进一步研究。
3.2mTOR与突触可塑性关键分子NMDA受体亚基磷酸化水平与LID发病机制
DR和NMDA受体的功能性相互作用已被广泛研究。众多的研究表明,活化D1R或D2R的可调节NMDA受体的功能和转运,例如,在纹状体长时程增强(LTP)的诱导要求D1R的活化,D1R拮抗剂阻断防止NMDA受体依赖性LTP,而这种效果是通过D1R的激活逆转[21,22]。NMDAR的NR1亚基由单个基因的8个剪接变异体,NMDAR通过其NR1-C末端区域NR1-C2和NR1-C2拼接的丰富程度与D1R相互作用。NMDAR聚合物与D1R内化诱导激动剂刺激相互作用,ERK的磷酸化(即激活)被限制于中型棘状神经元D1R的表达,并取决于D1R和谷氨酸NMDAR的伴随刺激,然而负责该激活的D1R和NMDAR各自的发生的机制仍未明确[23]。D1R激动剂可以增加NR2B(Y1472)的磷酸化水平[24]。触发NMDA受体的开放或关闭通常要受多种因子的影响,NMDA受体的磷酸化是其调控的最重要的机制之一[25]。
D1R和D2R激动剂-L-DOPA产生的运动障碍和类似的发病率已经明确并具有严重性,而基底节谷氨酸传输信号通路异常已被证明是在PD和LID的发展的重要参与者[26,27]。激动剂D1R会增加整体NMDA受体NR1亚单位的磷酸化水平[28]。NR2A和NR2B积极的参与了突触可塑性中的活动,置换NR2B与NR2A导致降低神经元突触的LTP[29],与D1R互动进一步增加了NMDAR系统的复杂性。D1R的激活是对谷氨酸(NMDA、AMPA等)诱导的长时程增强的皮质纹状体突触的生成至关重要,黑质纹状体多巴胺能投射到皮质纹状体接收运动信息的处理是非常重要的,在6-OHDA损毁大鼠中显著改变了纹状体的兴奋传导,该途径会导致类似于一些PD的症状等自发的变化[23]。从纹状体切片皮质纤维高频刺激(HFS)产生的LTP中突触电位会发生改变,兴奋性突触后电位(EPSP)通过刺激皮质纤维诱发LTP,可能观察到一些在PD中所表现的运动障碍[23,30]。谷氨酸(NMDA等)的动力学与D1R信号传导级联密切相关,多巴胺受体激动剂会促进纹状体中的NMDA亚基NR1(Ser896)的磷酸化表达[31]。多巴胺D1R耦合PKA的活化诱导NMDA受体NR1磷酸化,在中枢神经系统中丝氨酸896位点的磷酸化较为常见[32]。在纹状体神经元中可通过NR1亚基去磷酸化水平下调谷氨酸的活性[33]。慢性左旋多巴治疗6-OHDA所致的PD大鼠模型中会影响NMDA受体各亚基成分的变化,而且NMDAR1磷酸化表达水平升高[34]。Ser896是在NR1亚基上的胞内羧基端磷酸化位点上,该位点的磷酸化能够调节NMDA受体的活性和易化突触传递的作用。而NMDA受体的NR2A与NR2B两个亚基在突触可塑性中起着关键因素。
NMDA受体的活性是通过NR1亚基磷酸化的调制[35]。NMDANR2A亚单位Y1325磷酸化位点是主要的酪氨酸残基之一[36],其磷酸化水平被认为是NMDA受体的活性的一个良好指标,是调节突触可塑性的关键步骤[37]。另外从突触可塑性作为其关键分子,即从NMDANR1(ser896)磷酸化水平反应NMDA受体细胞膜数量、NMDA受体的活性的一个良好指标NMDANR2A(Y1325)以及NMDA受体最重要的NMDANR2B(Y1472)出发,突触可塑性的许多分子,至于具体如何以怎么样的突触参数等去平衡纹状体NMDA受体亚基磷酸化水平并降低LID发生的风险,其尚需要进一步研究。在突触后膜致密区(PSD)中NR2B是发生酪氨酸磷酸化调节的主要亚单位,其中Tyr1472是最重要的磷酸化靶位点[38,39]。
本研究发现,LID组的NMDA亚基磷酸化水平NR1(ser896)、NR2A(Y1325)、NR2B(Y1472)的表达水平均为最高,其中在LID组和C组之间两者比较无明显差异(P>0.05),雷帕霉素干预后NMDA亚基磷酸化水平NR1(ser896)、NR2A(Y1325)、NR2B(Y1472)的表达水平明显下降(P<0.05)。同样,在实验大鼠LID的模型中Gardoni等[40]研究发现在LID的发展过程中证实突触NMDA受体通过使用一种细胞渗透性肽的组合物来调制NR2A亚基以致减少的大鼠LID的进程中所占比,也就是说预防含NR2A-NMDA受体的突触异常激活足以确定具有一个显著减少LID发病的作用;靶向离子型谷氨酸受体剂可改善PD的运动症状,以及降低LID的运动行为的发作[41]。NMDA受体过度活化并与LID紧密关联,同样选择性NR2B亚基拮抗剂可以改善LID[42]。mTOR信号通路在突触水平的失调有牵连PD的发病机制,在突触水平适当的操纵AMPK/AKT/mTOR的信号是预防和治疗PD的一个潜在策略[43]。
本实验中也发现这一系列的突触关键分子磷酸化水平的变化均与mTOR信号有关,这可能是mTOR信号调控底物的磷酸化水平改变了MDA亚基NR1(ser896)、NR2A(Y1325)、NR2B(Y1472)磷酸化的表达水平,提示了激活mTOR信号通路可能不仅依赖PSD-95同时也依赖于关键分子NMDA受体亚基磷酸化一系列的突触活动引起LID。
因此本研究发现:1、mTORC1参与了LID的发病,应用mTOR特异性抑制剂雷帕霉素能够缓解异动症严重程度;雷帕霉素并不影响L-DOPA对PD的治疗,这与以往的研究相符[44];2、大鼠纹状体区的mTOR信号通路激活与LID的发生密切,D1R受体过度活化,mTOR信号的激活对纹状体的突触分子具有调节作用,导致突触可塑性的骨架分子PSD-95以及关键分子NMDA亚基的磷酸化表达水平升高,mTOR信号转导可能依赖突触分子PSD-95、NMDA受体NR1(ser896)、NR2A(Y1325)、NR2B(Y1472)亚基磷酸化等的表达引起突触活动过表达,使突触可塑性发生适应性改变,最终产生和维持皮质纹状体突触“病理性LTP”,出现基底节信号异常,从而介导了LID的发生。

图7 mTOR信号通路被激活管理L-DOPA及参与LID
总之,L-DOPA诱发运动障碍的突触机制,是悬而未决的问题[45]。本研究发现,从突触可塑性的一些分子水平表达当中得出mTOR信号通路激活异动症组中大鼠PSD-95、NMDA受体亚基的磷酸化表达水平升高,经特异性抑制mTOR信号后可以减轻LID症状并降低PSD-95、NMDA受体亚基的磷酸化表达水平,mTORC1参与LID的发病过程,而这过程中PSD-95、NMDA受体功能增强在“病理性LTP”产生中起重要作用,但是“病理性LTP”中的调控涉及到PSD-95、NMDA受体等到底是怎么整合的具体机制目前尚不清楚,有待进一步研究。
参考文献
[1]Calne DB,Reid JL,Vakil SD,et al.Idiopathic Parkinsonism treated with an extracerebral decarboxylase inhibitor in combination with levodopa[J].Br Med J,1971,3(5777):729-732.
[2]Santini E,Heiman M,Greengard P,et al.Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA-induced dyskinesia[J].Sci Signal,2009,2(80):ra36.
[3]Subramaniam S,Napolitano F,Mealer RG,et al.Rhes,a striatal-enriched small G protein,mediates mTOR signaling and L-DOPA-induced dyskinesia[J].Nat Neurosci,2011,15(2):191-193.
[4]Decressac M,Bj rklund A.mTOR Inhibition Alleviates L-DOPA-Induced Dyskinesia in Parkinsonian Rats[J].J Parkinsons Dis,2013,3(1):13-17.
[5]Neves G,Cooke SF,Bliss TV.Synaptic plasticity,memory and the hippocampus:a neural network approach to causality[J].Nat Rev Neurosci,2008,9(1):65 75.
[6]Picconi B,Centonze D,H kansson K,et al.Loss of bidirectional striatal synaptic plasticity in L-DOPA induced dyskinesia[J].Nat Neurosci,2003,6(5):501-506.
[7]Jenner P.Molecular mechanisms of L-DOPA-induced dyskinesia[J].Nat Rev Neurosci,2008,9(9):665-677.
[8]Torres EM,Lane EL,Heuer A,et al.Increased efficacy of the 6-hydroxydopamine lesion of the median forebrain bundle in small rats,by modification of the stereotaxic coordinates[J].J Neurosci Methods,2011,200(1):29-35.
[9]Paxinos G,Watson C.The rat brain in stereotaxic coordinates:hard cover edition[M].Academic press,2006.
[10]Thomas J,Wang J,TakubaH,et al.A 6-hydroxydopamine-induced selective parkinsonian rat model:further biochemical and behavioral characterization[J].Exp Neurol,1994,126(2):159-167.
[11]Monville C,Torres EM,Dunnett SB.Validation of the l-dopa-induced dyskinesia in the 6-OHDA model and evaluation of the effects of selective dopamine receptor agonists and antagonists[J].Brain Res Bull,2005,68(1):16-23.
[12]Zhang J,Saur T,Duke AN,et al.Motor Impairments,Striatal Degeneration,and Altered Dopamine-Glutamate Interplay in Mice Lacking PSD-95[J].J Neurogenet,2014,28(1-2):98-111.
[13]Kim E,Sheng M.PDZ domain proteins of synapses[J].Nat Rev Neurosci,2004,5(10):771-781.
[14]Scannevin RH,Huganir RL.Postsynaptic organisation and regulation of excitatory synapses[J].Nat Rev Neurosci,2000,1(2):133-141.
[15]El-Husseini AE,Schnell E,Chetkovich DM,et al.PSD-95 involvement in maturation of excitatory synapses[J].Science,2000,290(5495):1364-1368.
[16]Porras G,Berthet A,Dehay B,et al.PSD-95 expression controls L-DOPA dyskinesia through dopamine D1 receptor trafficking[J].J Clin Invest,2012,122(122 (11)):3977-3989.
[17]Kim MJ,Futai K,Jo J,et al.Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95[J].Neuron,2007,56(3):488-502.
[18]Ehrlich I,Malinow R.Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity[J].J Neurosci,2004,24(4):916-927.
[19]Stein V,House DR,Bredt DS,et al.Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression[J].J Neurosci,2003,23(13):5503-5506.
[20]Ba M,Kong M,Ma G.Postsynaptic density protein 95-regulated nr2B tyrosine phosphorylation and interactions of Fyn with nr2B in levodopa-induced dyskinesia rat models[J].Drug Des Devel The,2014,19(9):199-206.
[21]Calabresi P,Gubellini P,Centonze D,et al.Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation,opposing forms of synaptic plasticity[J].J Neurosci,2000,20(22):8443-8451.
[22]Kerr JN,Wickens JR.Dopamine D-1/D-5 receptor activation is required for long-term potentiation in the rat neostriatum in vitro[J].J Neurophysiol,2001,85(1):117-124.
[23]Fiorentini C,Gardoni F,Spano PF,et al.Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-D-aspartate receptors[J].J Biol Chem,2003,278(22):20196-20202.
[24]Pascoli V,Besnard A,Herv D,et al.Cyclic adenosine monophosphate-independent tyrosine phosphorylation of NR2B mediates cocaine-induced extracellular signal-regulated kinase activation[J].Biol Psychiatry,2011,69(3):218-227.
[25]Chen BS,Roche KW.Regulation of NMDA receptors by phosphorylation[J].Neuropharmacology,2007,53(3):362-368.
[26]Gerfen CR,Surmeier DJ.Modulation of striatal projection systems by dopamine[J].Annu Rev Neurosci,2011,34:441-466.
[27]Bellone C,Gardoni F.Modulation of the glutamatergic transmission by Dopamine:a focus on Parkinson,Huntington and Addiction Diseases[J].Front Cell Neurosci,2015,9:25.
[28]Snyder GL,Fienberg AA,Huganir RL,et al.A dopamine/D1 receptor/protein kinase A/dopamine-and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor[J].J Neurosci,1998,18(24):10297-10303.
[29]Barria A,Malinow R.NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII[J].Neuron,2005,48(2):289-301.
[30]Lee FJ,Xue S,Pei L,et al.Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor[J].Cell,2002,111(2):219-230.
[31]Lee DK,Bian S,Rahman MA,et al.Repeated cocaine administration increases N-methyl-d-aspartate NR1 subunit,extracellular signal-regulated kinase and cyclic AMP response element-binding protein phosphorylation and glutamate release in the rat dorsal striatum[J].Eur J Pharmacol,2008,590(1-3):157-162.
[32]Dudman JT,Eaton ME,Rajadhyaksha A,et al.Dopamine D1 receptors mediate CREB phosphorylation via phosphorylation of the NMDA receptor at Ser897 NR1[J].J Neurochem,2003,87(4):922-934.
[33]Choe ES,Shin EH,Wang JQ.Inhibition of protein phosphatase 2B upregulates serine phosphorylation of N-methyl-D-aspartate receptor NR1 subunits in striatal neurons in vivo [J].Neurosci Lett,2005,384(1-2):38-43.
[34]Dunah AW,Wang Y,Yasuda RP,et al.Alterations in Subunit Expression,Composition,and Phosphorylation of StriatalN-Methyl-d-Aspartate Glutamate Receptors in a Rat 6-Hydroxydopamine Model of Parkinson's Disease[J].Mol Pharmacol,2000,57(2):342-352.
[35]Masu M,Nakajima Y,Moriyoshi K,et al.Molecular characterization of NMDA and metabotropic glutamate receptors[J].Ann N Y Acad Sci,1993,707(1):153-164.
[36]Krupp JJ,Vissel B,Thomas CG,et al.Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors[J].Neuropharmacology,2002,42(5):593-602.
[37]Grosshans DR,Clayton DA,Coultrap SJ,et al.LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1[J].Nat Neurosci,2001,5(1):27-33.
[38]Moon IS,Apperson ML,Kennedy MB.The major tyrosine-phosphorylated protein in the postsynaptic density fraction is N-methyl-D-aspartate receptor subunit 2B[J].Proc Natl Acad Sci USA,1994,91(9):3954-3958.
[39]Nakazawa T,Komai S,Tezuka T,et al.Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor[J].J Biol Chem,2001,276(l):693一699 .
[40]Gardoni F,Sgobio C,Pendolino V,et al.Targeting NR2A-containing NMDA receptors reduces L-DOPA-induced dyskinesias[J].Neurobiol Aging,2012,33(9):2138-2144.
[41]Gardoni F,Di Luca M.Targeting glutamatergic synapses in Parkinson's disease[J].Curr Opin Pharmacol,2015,20:24-28.
[42]Kong M,Ba M,Liu C,et al.NR2B antagonist CP-101,606 inhibits NR2B phosphorylation at tyrosine-1472 and its interactions with Fyn in levodopa-induced dyskinesia rat model[J].Behav Brain Res,2015,286:46-53.
[43]Xu Y,Liu C,Chen S,et al.Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson's disease[J].Cell Signal,2014,26(8):1680-1689.
[44]Decressac M,Bj rklund A.mTOR inhibition alleviates L-DOPA-induced dyskinesia in parkinsonian rats[J].J Parkinsons Dis,2013,3(1):13-17.
[45]Cenci MA.Presynaptic mechanisms of L-DOPA-induced dyskinesia:the findings,the debate,and the therapeutic implications[J].Front Neurol,2014,5:242.
(2015-11-12收稿2016-04-12修回)
基金项目:国家自然科学基金资助项目(项目编号为31260241)
【中图分类号】R
【文献标识码】A
【文章编号】1007-0478(2016)03-0182-09
