大前庭水管综合征家系特征与SLC26A4基因分析
2016-06-30李伍高严提珍曾定元唐宁李哲涛唐向荣覃文华罗世强杨艳柳州市出生缺陷预防与控制重点实验室广西柳州55002柳州市妇幼保健院医学遗传科广西柳州5500柳州市妇幼保健院听力诊断中心广西柳州5500柳州市妇幼保健院放射科广西柳州5500
李伍高严提珍曾定元唐宁李哲涛唐向荣覃文华罗世强杨艳柳州市出生缺陷预防与控制重点实验室(广西柳州5500)2柳州市妇幼保健院医学遗传科(广西柳州5500)柳州市妇幼保健院听力诊断中心(广西柳州5500)柳州市妇幼保健院放射科(广西柳州5500)
大前庭水管综合征家系特征与SLC26A4基因分析
李伍高1,2严提珍1,2曾定元1唐宁1,2李哲涛1,2唐向荣3覃文华4罗世强1,2杨艳3
1柳州市出生缺陷预防与控制重点实验室(广西柳州545001)
2柳州市妇幼保健院医学遗传科(广西柳州545001)
3柳州市妇幼保健院听力诊断中心(广西柳州545001)
4柳州市妇幼保健院放射科(广西柳州545001)
【摘要】目的分析一个大前庭水管综合征家系的临床特征和SLC26A4基因检测特点。方法对一个大前庭水管综合征家系进行病史采集和听力学检测,绘制耳聋家系系谱图,提取受检者的基因组DNA,对所有家系成员进行耳聋基因芯片分析和SLC26A4基因全外显子及外显子侧翼序列的检测。结果该家系共5代合计30人(男17人,女13人),现存26人,其中第四代7人为耳聋患者,6人均为语前感音神经性聋。1人为语后感音神经性聋,颞骨CT显示均为前庭水管扩大,第五代1人为耳聋患者,表现为前庭水管扩大合并语前感音神经性聋。在家系中共发现SLC26A4基因c.754C>T、c.919-2A>G、c.1264-12T>A和c.1548_1549insC四种已知致病突变类型。耳聋患者均为复合杂合突变。10人为SLC26A4基因携带者。结论该家系的8例耳聋患者由SLC26A4基因复合杂合突变导致前庭水管扩大,病因学分析可为患者提供预见性的临床措施,并为该家系的后代遗传咨询与婚育指导提供理论依据及科学手段,有效地防止聋儿后代出生。
【关键词】前庭导水管扩大综合征;SLC26A4基因;基因芯片;Sanger测序;基因突变
Funding:This work was funded by a grant from the National Natural Science Fund of China(NSFC,No.81360159),a grant from the Technology Research and Development program of Guangxi(No.14124004-1-20),a grant from Liuzhou Science and Technology Development Funds(No.2014J030401)and a grant from the Technology Research and Development program of Liuzhou(No.2014G020404).
Competing Interests:The authors have declared that no competing interests exist.
大前庭水管综合征(large vestibular aqueduct syndrome,LVAS)是上世纪70年代末随CT问世而发现的遗传性耳聋患儿中最常见的一种内耳发育畸形性疾病,也是一种常染色体隐性遗传性听力障碍性疾病[1]。主要表现为前庭水管扩大合并感音神经性或混合性耳聋,一般不伴有其他内耳发育异常和其他器官系统的异常。LVAS的致病基因定位在人类染色体7q31的SLC26A4基因上,该基因编码Pendrin蛋白,可导致Pendred综合征[2,3]。本研究应用耳聋基因芯片技术联合Sanger测序法对一个非综合征性大前庭水管综合征家系进行SLC26A4基因检测,分析SLC26A基因的遗传特征。
1 资料与方法
1.1家系调查方法
本研究家系的调查分析已获得柳州市妇幼保健院伦理委员会的伦理认证及认可。家系(编号E40 和E44)位于广西柳州市柳城县东泉镇,两个家系有亲属关系,因此合并为大家系进行分析。先证者1和先证者2于2013年8月就诊于柳州市妇幼保健院听力诊断中心,分别进行了纯音测听、声导抗、听性脑干反应、DPOAE及颞骨CT扫描检查,该家系成员无明确的耳毒性药物应用史及噪声接触史,不伴其他器官系统异常。2013年9月对该家系所有受检者进行了详细的病史询问、严格的专科检查及听力学检测。耳聋的表型判断标准依据王秋菊[4]编译的《关于非综合征型遗传性听损伤家系遗传学及听力学描述术语建议案》。根据Valvassori等[1]的颞骨CT诊断标准:前庭水管的外口与总脚或峡部间中点处直径大于1.5 mm即为前庭水管扩大。
1.2DNA提取和浓度测定
采取受检对象及家系成员的外周静脉血3~5 ml(EDTA.K2抗凝)。利用试剂盒(北京天根生化科技有限公司)提取外周血基因组DNA,提取步骤参照试剂盒使用说明进行。利用ASP-2680微量分光光度计(美国ACTGene公司)对样本的基因组DNA的提取质量和浓度进行检测。DNA样本的OD260nm/ OD280nm比值应在1.6~2.0之间,OD260nm/ OD230nm比值需≥2.0,浓度需≥50 ng/L。
1.3耳聋基因芯片检测
应用遗传性耳聋基因芯片检测试剂盒(北京博奥生物有限公司)对GJB2基因的c.35delG、c.176del16、c.235delC、c.299delAT,GJB3基因的c.538C>T,SLC26A4基因的c.919-2 A>G、c.2168A>G,线粒体12S rRNA基因的m.1494C>T、m.1555A>G等4个耳聋基因共9个突变位点进行检测。设备为晶芯LuxS-canTM10K-A微阵列芯片扫描仪及配套软件。
1.4DNA测序检测
采用Sanger测序针对SLC26A4进行序列分析。引物序列见参考文献[5],PCR产物行1.5%琼脂糖凝胶电泳,选取PCR扩增产物纯化,测序标本外送广州立菲测序有限公司进行检测。
1.5序列比对分析
Sanger测序结果用DNAStar软件包中的Seqman软件与NCBI网站(http://www.ncbi.nlm.nih.gov/)公布的SLC26A4标准序列(NC_000007.13)进行序列比对分析。
2 结果
2.1家系系谱图和听力学特征
家系系谱图见图1,该家系共5代合计30人(男17人,女13人),现存26人,年龄介于4~77岁之间,共8人患双耳感音神经性耳聋。先证者1(Ⅳ:3)4岁,先证者2(V:1)2岁,均于出生时进行了听力筛查,初筛和复筛结果均未通过,纯音听阈测试显示双耳对称性极重度感音神经性聋,听阈曲线呈下降型,颞骨CT扫描结果显示前庭水管扩大(见图2A和2B)。先证者1(Ⅳ:3)的父母听力正常,先证者2(V:1)的父母均为前庭水管扩大合并双耳对称性极重度感音神经性耳聋患者(Ⅳ:8和Ⅳ:9)。患者(Ⅳ:1、Ⅳ:2、Ⅳ:7、Ⅳ:8、Ⅳ:9)均于出生时发病,均表现为双耳对称性极重度感音神经性耳聋,听阈曲线双耳对称均为下降型,颞骨CT扫描结果均显示前庭水管扩大。患者(Ⅳ:10)于14岁出现听力下降,随后表现为双耳对称性进行性加重的重度感音神经性耳聋,CT扫描结果也显示前庭水管扩大。其他家庭成员听力正常。从系谱图可见,该家系符合常染色体隐性遗传性耳聋特征。
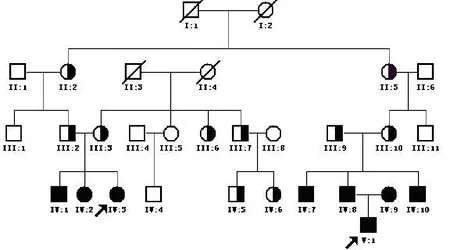
图1 耳聋家系系谱图Fig.1 Pedigree of deafness family
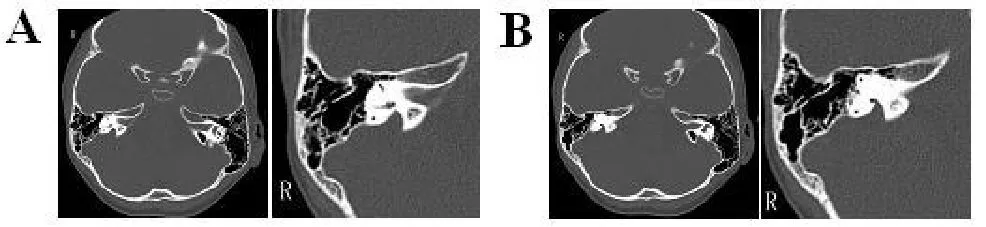
图2 大前庭水管颞骨CT示意图A:患者IV:3颞骨CT示意图(右侧);B:先证者V:1颞骨CT示意图(右侧)。Fig.2 The CT scaning of LVAS Patients.A:The CT diagram of patient IV:3;B:The CT diagram of proband V:1.
2.2耳聋基因芯片的检测结果
基因芯片法检出患者(Ⅳ:9)携带SLC26A4基因c.919-2A>G单杂合突变,先证者和其他家系成员未发现突变。患者(Ⅳ:9)的基因芯片图谱见图3。
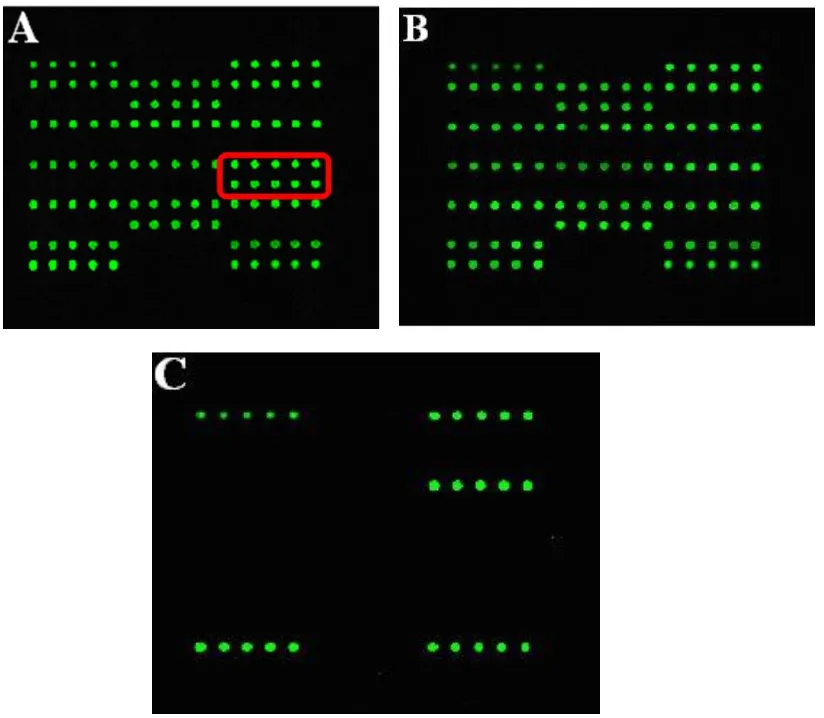
图3 患者(Ⅳ:9)的基因芯片结果A:c.919-2A>G/WT;B:正常对照;C:空白对照(SBC)Fig.3 Gene chip profiling in a deafness patient(Ⅳ:9).A:c.919-2A>G carrier;B:Normal control;C:Sample Blank Control(SBC).
2.3SLC26A4基因Sanger测序结果
因先证者和其他患者颞骨CT结果均显示为前庭水管扩大综合征患者,基因芯片筛查常见的SLC26A4基因c.919-2 A>G和c.2168A>G突变,仅在患者(Ⅳ:9)发现单杂合突变,为明确病因,本研究设计引物,通过PCR扩增及Sanger测序对SLC26A4基因全外显子进行了检测。结果发现先证者1(Ⅳ:3)为c.1264-12T>A和c.1548_1549insC复合杂合突变,其哥哥和姐姐均为耳聋患者,其基因型与Ⅳ:3相同。Ⅱ:2、Ⅲ:2、Ⅲ:3、Ⅲ:6、Ⅲ7、Ⅳ:5和Ⅳ:6为SLC26A4基因突变携带者,Ⅱ:1、Ⅱ:4、Ⅱ:5、Ⅲ:1、Ⅲ:8、Ⅳ:4基因型正常。先证者2(V:1)为c.754T>C和c.1548_1549insC复合杂合突变,其父亲(IV:7)基因型与V:1相同,其母亲(Ⅳ:8)为c.919-2A>G和c.1548_1549insC复合杂合突变。Ⅳ:7、Ⅳ:10的基因型与Ⅳ:8相同。Ⅱ:5、Ⅲ:9、Ⅲ:10均为SLC26A4基因突变携带者,Ⅱ:6和Ⅲ:11基因型正常。家系各成员基因检测结果见表1,基因突变的测序结果见图4。
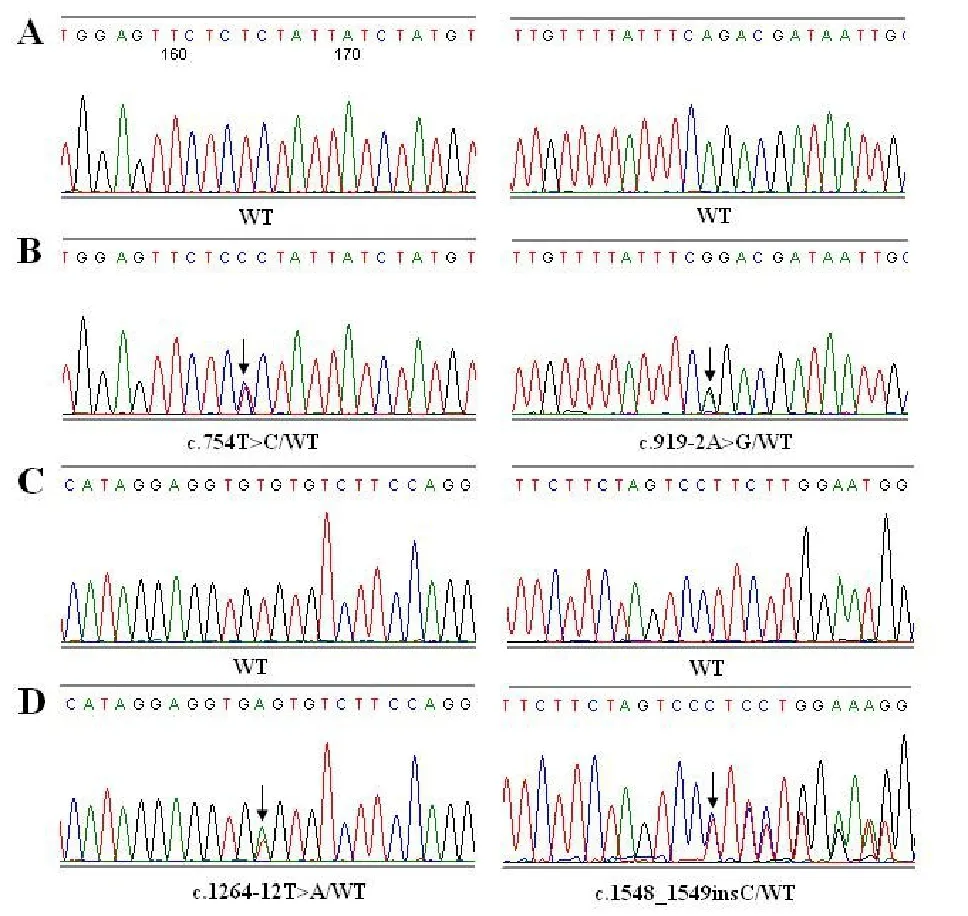
图4 SLC26A4基因Sanger测序图谱Fig.4 Sanger sequencing chromatograms of the SLC26A4 gene.WT represents wild type.Arrow indicates the position of mutation.
3 讨论
大前庭水管综合征的听力学特点多表现为双侧进行性感音神经性耳聋,CT或MRI提示前庭水管或内淋巴囊扩大[6],不同个体在出生时听力损伤程度具有较大差别,从听力完全正常至极重度听力障碍,头部外伤、剧烈体育活动、头部倒立、感冒、用力擤鼻或咳嗽等可造成明显听力下降。早期诊断并采取正确防护措施,有助于保留患者的残余听力,维持生活质量。对于本研究家系发现的8例耳聋患者,其中7例患者自出生后即对声音反应差,表现为言语障碍,纯音测听及听性脑干反应检测结果提示极重度感音神经性耳聋;1例(Ⅳ:10)出生至14岁前听力正常,14岁加入学校篮球队参加训练后,发现听力逐渐下降,随后发展为双耳进行性极重度感音神经性耳聋。全部患者高分辨率颞骨CT显示前庭水管扩大,未合并其它内耳畸形。体格检查结果未发现颈部肿大,甲状腺功能均正常。对于Ⅳ:10患者若在其出生时、入学前或最初发生听力损失的时候进行耳聋基因诊断或影像学检查,早期确诊,就可能通过改变生活模式,加强听觉防护等措施延缓或防止听力下降,获得更好的生活质量。
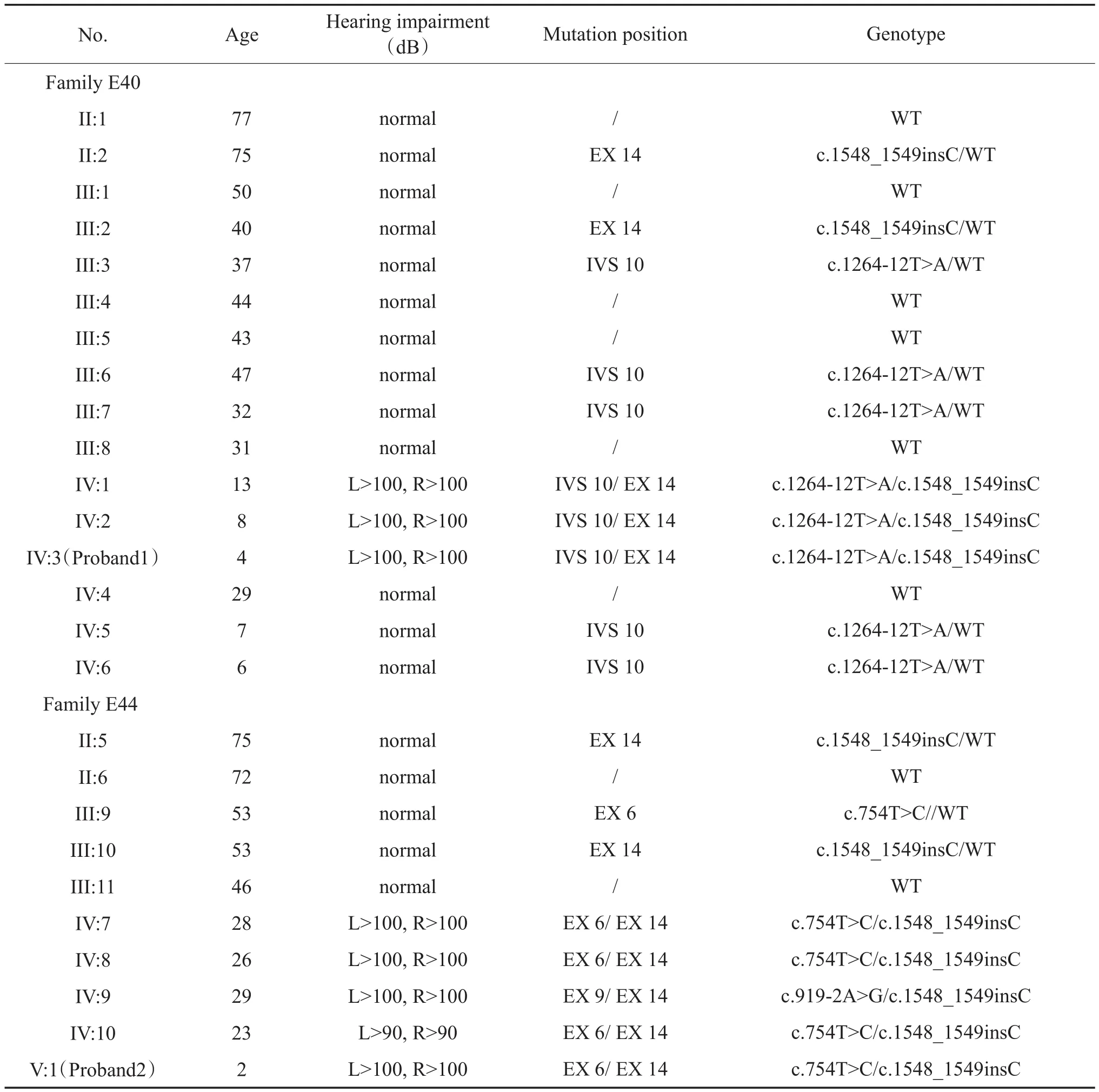
表1 家系成员临床表型和基因型Table 1 Clinical phenotype and genotype of family members
1999年,Usami等[5]把大前庭水管综合征的基因定位在7q31(即SLC26A4基因),其包括21个外显子(1号外显子不参与编码),mRNA全长为4930 bp,开放阅读框架为2343 bp。SLC26A4基因编码的蛋白为Pendrin蛋白,由780个氨基酸组成,主要表达于内耳的内淋巴囊和淋巴管,介导Cl-、HCO3-、I-的转运,维持内淋巴液的离子平衡,对内耳内淋巴液的再吸收具有重要的作用[7]。SLC26A4基因突变可导致Pendred综合征和呈常染色体隐性遗传的非综合征性聋DFNB4。目前报道的SLC26A4基因的突变位点已超过240个(http://www.healthcare.uiowa.edu/labs/pendredandbor/slcMutations.htm),其中绝大部分是错义突变(占86%左右),另外还有剪接位点突变和移码突变等。这些突变在mRNA链上的分布无明显聚集现象。SLC26A4基因的突变在LVAS病人中检出率我国为92.1~95.4%[7]。SLC26A4基因具有广泛的遗传异质性,研究发现SLC26A4基因在中国正常人群中有较高的携带率为3%,在中国耳聋人群中c.919-2A>G和c.2168A>G是热点突变[8]。
本研究利用基因芯片联合Sanger测序技术通过对一个大家系中8例耳聋患者及18例正常听力家系成员进行SLC26A4基因检测,发现c.1548_1549insC、c.1264-12T>A、c.919-2A>G、c.754T>C四种突变。这四种突变在既往文献中已有报道,其致病性明确,且c.919-2A>G(ⅣS7-2A>G)为中国人群中的热点突变,其它三种为非热点突变。c.1548_1549insC(p.S517fs)移码突变使终止密码子提前出现,产生仅有525个氨基酸的截短蛋白,影响pendrin蛋白的结构和功能[9];2007年国内学者Hu等[10]首次报道了c.1264-12T>A(ⅣS10-12T>A)突变,该突变导致11号外显子的丢失,使外显子10和12直接相连,随后相继有研究团队在湖南地区大前庭水管综合征患者中也发现该突变[11],目前该突变只在中国人群中报道过,其他人群尚未见报道;c.919-2A>G突变位点位于外显子8剪接位点的位置(即内含子7的3'末端),它的突变使前体mRNA无法正常剪接,整个外显子8丢失,外显子7和9直接相连,从而导致pendrin的翻译发生移码或提前终止,影响pendrin蛋白的结构和功能[9];中国解放军总医院戴朴研究团队在全国耳聋分子流行病学调查中发现c.919-2 A>G在耳聋患者中的检出率高达12.69%,该突变频率占该基因总突变的57.63%[8];c.754T>C(p.S252P)为错义突变,导致丝氨酸改变为脯氨酸,最终影响pendrin蛋白的结构和功能[9]。
本研究发现8名患者携带SLC26A4双等位基因的复合杂合突变,经颞骨高分辨CT诊断均为LVAS患者;10名成员携带SLC26A4单个等位基因突变,均为听力正常者。家系遗传图谱显示,第四代(Ⅳ:1、Ⅳ:2和Ⅳ:3)基因检测结果均为c.1264-12T>A和c.1548_ 1549insC的复合杂合突变,其父母(Ⅲ:2和Ⅲ:3)听力正常,但分别是c.1548_1549insC和c.1264-12T>A的突变携带者;第四代(Ⅳ:7、Ⅳ:8和Ⅳ:10)基因检测结果均为c.754T>C和c.1548_1549insC的复合杂合突变,其父母(Ⅲ:9和Ⅲ:10)听力正常,但分别是c.754T>C和c.1548_1549insC的突变携带者。第五代(V:1)基因检测结果为c.754T>C和c.1548_1549insC复合杂合突变,其父母均为耳聋患者,父亲是c.754T>C和c.1548_1549insC复合杂合突变,母亲为c.919-2A>G 和c.1548_1549insC复合杂合突变,V:1的c.754T>C突变等位基因来自父亲,c.1548_1549insC突变等位基因来自母亲。由此可见,SLC26A4突变的等位基因会稳定地由亲代向子代传递。
目前大部分基层医疗机构和医务人员尚未充分认识防聋治聋工作的紧迫性和重要性,聋哑人之间婚配比例较高,婚配生育缺少医疗机构的系统有效的指导和干预;同时由于感音神经性耳聋的不可逆性及治疗的高成本性,目前临床医务人员的工作重点为如何实现耳聋的预防及早期干预。SLC26A4基因突变是LVAS的根本性致病因子,遵循常染色体隐性遗传规律,这为临床开展LVAS的基因诊断和产前诊断提供了理论依据[12]。产前诊断对于已生育过大前庭水管综合征聋儿的家庭来说,超过70%的家庭可以通过基因诊断明确病因,并将再次生育聋儿的风险由25%降低到1~3‰,从而避免因聋儿出生而带来沉重的负担。为达到对耳聋的一级预防,需要对家系中听力正常成员进行SLC26A4基因的筛查,对于筛查查出的携带者(如Ⅳ:5和Ⅳ:6),婚育前需要进一步对其配偶进行SLC26A4基因的筛查。本家系患者(Ⅳ:1、Ⅳ:2、Ⅳ:3、Ⅳ:7、Ⅳ:10和V:1)在婚育前,需对其配偶进行耳聋基因诊断,以避免因后者也携带SLC26A4基因突变,而使他们生育聋儿的几率高达50%以上。值得注意的是先证者(V:1)的父亲为LVAS患者(基因型为c.754T>C/c.1548_1549insC),选择了同为LVAS患者的女性(基因型为c.919-2A>G/c.1548_1549insC)结婚,根据孟德尔遗传规律,该家庭生出大前庭水管综合征患儿的风险率为100%,无法通过产前诊断或植入前产前诊断(preimplantation genetic diagnosis,PGD)再生育一个听力正常的小孩,只能通过早干预、早治疗来提高患者的生活质量。此外,基因诊断可先于影像学检查诊断LVAS患者,减少婴幼儿患者早期暴露子CT检查放射线辐射的风险。目前,使重度感音神经性耳聋患者回到有声世界的最好办法是人工耳蜗植入术,由于SLC26A4突变主要影响内耳的功能,这提示LVAS患者将会有良好的人工耳蜗康复效果。
至今发现SLC26A4基因突变具有明显的异质性。其突变的种类众多,且不同种族人群的SLC26A4基因突变图谱不相同,不同的种族找到SLC26A4双等位基因的比率亦不相同。研究发现约有10~20%的LVAS患者只能筛查到SLC26A4单等位基因突变或未发现任何突变,可能的原因是SLC26A4基因的启动子区域或者具有潜在剪接位点的内含子区存在突变,也可能与该基因拷贝数变异有关[13,14]。随着检测技术的普及和检测成本的下降,高通量测序是未来检测遗传性耳聋基因的必然趋势。
综上所述,耳聋基因诊断可以在分子水平明确大前庭水管综合征的病因,开展遗传咨询、婚育指导、耳聋预测及出生缺陷的干预。随着测序分析准确性高,灵敏性强,且成本越来越低,自动化程度越来越高,通量越来越大,可实现对先证者及其家系成员的SLC26A4基因突变位点的快速筛查,确诊患者及携带者,为该家系及其同类事件的发生提供正确而准确的临床医学指导。在患者将来婚配时对其配偶进行相应位点基因检测,通过产前耳聋基因检测来指导生育,以预防其生育耳聋后代,达到耳聋的一级预防,减轻社会及家庭的负担。
参考文献
1Valvassori GE,Clemis JD.The large vestibular aqueduct syndrome [J].Laryngoscope,1978,88(5):723-728.
2Ito T,Choi BY,King KA,et al.SLC26A4 genotypes and phenotypes associated with enlargement of the vestibular aqueduct[J].Cell Physiol Biochem,2011,28(3):545-552.
3Soh LM,Druce M,Grossman AB,et al.Evaluation of genotype-phenotype relationships in patients referred for endocrine assessment in suspected Pendred syndrome[J].Eur J Endocrinol,2015,172(2):217-226.
4王秋菊,顾瑞.关于非综合征型遗传性听损伤家系遗传学及听力学描述术语建议案[J].中华耳科学杂志,2003,1(4):46-47,67.Qiuju Wang,Rui Gu.Recommendations for the Description of Genetic and Audiological Data for Families with Nonsyndromic Hereditary Hearing Impairment[J].Chinese Journal of Otology,2003,1(4):46-47,67.
5Usami S,Abe S,Weston MD,et al.Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations[J].Hum Genet,1999,104(2):188-192.
6孙宝春,代志瑶,黄莎莎等.GJB2、SLC24A4基因致病性突变与内耳CT表型关系的研究[J].中华耳科学杂志.2014,12(1):30-33.Baochun Sun,Zhiyao Dai,Shasha Huang,et al.Study on the Relationship between the Pathogenic Mutations of GJB2、SLC26A4 and CT Phenotypes of Inner Ear in Patient with Sensorineural Hearing Loss[J].Chinese Journal of Otology,2014,12(1):30-33.
7赵亚丽,瞿所强,王秋菊.大前庭水管综合征及其相关的基因―SLC26A4[J].中华耳科学杂志,2005,3(4):289-292.Yali Zhao,Suoqiang Qu,Qiuju Wang.Large vestibul araqueduct syndrome and related gene—SLC26A4[J].Chinese Journal of Otology,2005,3(4):289-292.
8Dai P,Li Q,Huang D,et al.SLC26A4 c.919-2A>G varies among Chinese ethnic groups as a cause of hearing loss[J].Genet Med,2008,10(8):586-592.
9Park HJ,Lee SJ,Jin HS,et al.Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans[J].Clin Genet,2005,67(2):160-165.
10 Hu H,Wu L,Feng Y,et al.Molecular analysis of hearing loss associated with enlarged vestibular aqueduct in the mainland Chinese:a unique SLC26A4 mutation spectrum[J].J Hum Genet,2007,52(6):492-497.
11蒋璐,冯永,陈红胜等.中国湖南地区非综合征性聋患者SLC26A4基因突变研究[J].临床耳鼻咽喉头颈外科杂志,2010,24(13):587-591.Lu Jiang,Yong Feng,Hongsheng Chen,et al.An investigation of SLC26A4 gene mutation in nonsydromic hearing impairment in human province of China[J].Journal of Clinical Otorhinolarygology,2010,24(13):587-591.
12朱发梅,胡鹏,赖若沙等.基因芯片联合DNA测序法在大前庭水管综合征患者基因诊断中的应用[J].中华耳科学杂志,2011,9(2):168-173.Famei Zhu,Peng Hu,Ruosha Lai,et al.Application of DNA microarray and sequencing in genetic diagnosis of enlarged vestibular aqueduct syndrome[J].Chinese Journal of Otology,2011,9(2):168-173.
13赵建东,袁永一,王国建等.非综合征性前庭水管扩大患者拷贝数变异基因芯片筛查的分析[J].中华耳科学杂志,2014,12(1):23-25.Jiandong Zhao,Yongyi Yuan,Guojian Wang,et al.The analysis of Cytogenetics microarray screening in patients with non-syndromic Enlarged Vestibular Aqueduct[J].Chinese Journal of Otology,2014,12(1):23-25.
14Pang X,Chai Y,He L,et al.A 7666-bp genomic deletion is frequent in Chinese Han deaf patients with non-syndromic enlarged vestibular aqueduct but without bi-allelic SLC26A4 mutations[J].Int J Pediatr Otorhinolaryngol,2015,79(12):2248-2252.
Clinical features and gene analysis in a large Chinese family with large vestibular aqueduct syndrome
LI Wugao1,2,YAN Tizhen1,2,Zeng Dingyuan1,TANG Ning1,2,LI Zhetao1,2,TANG Xiangrong3,QIN Wenhua4,LUO Shiqiang1,2,YANG Yan3
1 Liuzhou Key Laboratory for Birth Defects Prevention and Control,Liuzhou,545001,China
2 Department of Medical Genetics,Liuzhou Maternity and Child Healthcare Hospital,Liuzhou,545001,China
3 Hearing Diagnosis Center,Liuzhou Maternity and Child Healthcare Hospital,Liuzhou,545001,China
4 Department of Radiology,Liuzhou Maternity and Child Healthcare Hospital,Liuzhou,545001,China
Corresponding author:YAN TizhenEmail:439078813@qq.com
【Abstract】Objective To report clinical and SLC26A4 assessment results of a large Chinese family with large vestibular aqueduct syndrome(LVAS).Methods The family tree was drawn based on medical history and audiological findings.Gene microarray and Sanger sequencing were performed using the genome DNA of the probands and other patients.The sequencing data were analyzed.Results There were 30 people(17 males and 13 females)in this family of 5 generations.Twenty-six were alive.There were 7 deaf patients in the fourth generation and 1 in the fifth generation.The clinical characteristics included pre- and post-lingual severe sensorineural hearing loss and enlarged vestibular aqueduct.A total of 4 known types of SLC26A4 mutations(c.754C>T,c.919-2A>G,c.1264-12T>A and c.1548_1549insC)were identified.Conclusion Hearing loss in the eight patients in this family is probably caused by various biallelic mutations of the SLC26A4 gene.Etiology analysis can predict clinical therapies required in these patients and provide a basis for marriage and repro-duction counseling.
【Key words】Large vestibular aqueduct syndrome(LVAS);SLC26A4 gene;Gene microarray;Sanger sequencing;Gene mutationidentified in the large family.
【中图分类号】R764
【文献标识码】A
【文章编号】1672-2922(2016)02-234-6
DOI:10.3969/j.issn.1672-2922.2016.02.022
基金项目:国家自然科学资金项目(81360159)、广西科技攻关项目(桂科攻14124004-1-20)、柳州市科技攻关项目(2014J030401)、柳州市科学研究与技术开发计划项目研究成果资助(2014G020404)
作者简介:李伍高,本科,技师,研究方向:遗传病的分子基础和基因诊断
通讯作者:严提珍,Email:439078813@qq.com
收稿日期:(2015-10-09审核人:冰丹)
