硫化亚铁矿物的生物合成及其对六溴环十二烷的还原脱溴研究
2016-06-23朱锡芬鲜海洋彭平安
毛 喆, 李 丹, 钟 音, 朱锡芬,鲜海洋, 彭平安
(1. 中国科学院 广州地球化学研究所 有机地球化学国家重点实验室, 广东 广州 510640; 2. 中国科学院 广州地球化学研究所中国科学院矿物学与成矿学重点实验室, 广东 广州 510640; 3. 广东省矿物物理与材料研究开发重点实验室, 广东 广州510640; 4. 中国科学院大学, 北京 100049)
0 引 言
六溴环十二烷(hexabromocyclododecane, HBCD)是目前世界上使用量最大的溴代阻燃剂之一, 广泛应用于建筑材料、纺织品以及电子产品中。据统计,1999年、2001年及2006年全球HBCD产量分别达到 15900 t、16700 t和 20000 t[1–2]。由于大量使用,HBCD在亚洲、欧洲、北美及北极地区等地的大气、水、土壤和沉积物等多种环境介质中被广泛检出[3–10]。而HBCD亲脂性强, 容易通过食物链或其他途径在生物体内富集, 具有生物毒性, 对人体和环境健康构成严重威胁。研究表明HBCD会影响哺乳动物的神经系统, 对肝脏有一定的毒性[10], 甚至有可能导致人体基因重组, 并进一步引起一系列疾病[11–12]。2013年联合国环境规划署将HBCD列入《关于POPs的斯德哥尔摩公约》禁用化学制品的黑名单[1]。因此, 如何有效控制和降低 HBCD的生态环境风险已成为一个亟待解决的科学问题。
由于HBCD疏水性强, 极易吸附和富集于环境土壤、沉积物、悬浮颗粒中, 其在土壤/沉积物中的环境行为引起了国内外研究人员的广泛关注[3–10]。HBCD在土壤/沉积物中的自然衰减包括生物降解与非生物转化, 是其环境行为的重要组成部分。已有的研究主要集中在 HBCD 的生物降解[13–17]。但是,在高污染、低温和酸性等极端环境胁迫下, 微生物生长和活性会受到抑制, 而非生物转化会成为HBCD自然衰减的重要途径。近年来有不少研究报道了 HBCD的热降解和光降解过程[18–21]; 但是, 关于环境中的自然氧化还原剂对HBCD的非生物转化研究十分薄弱。仅见Loet al.[22]报道了溶解态硫离子(被认为较为广泛地存在于厌氧的沉积物环境中)非生物转化HBCD的过程和机制。
硫化亚铁(FeS)矿物是一类重要的自然还原剂,广泛存在于土壤、河流沉积物、地下水、近海等环境中, 主要由硫酸盐还原菌(sulfate-reducing bacteria,SRB)通过利用环境中的有机质作为碳源和代谢能量,将硫酸盐还原成硫化氢, 硫化氢再与环境中的可溶性铁沉淀而形成的硫铁矿物。硫铁矿物在沉积物中的形态主要有: 无定形FeS、四方硫铁矿Fe0.995~1.023S、硫复铁矿 Fe3S4、黄铁矿 FeS2等[23]。大量研究表明硫铁矿物具有还原转化卤代有机污染物的活性, 能够将多种氯代和溴代有机污染物(如三氯乙烯、四氯乙烯、六氯乙烷、五氯乙烷、1, 1, 2, 2-和1, 1, 1, 2-四氯乙烷、1, 1, 1-三氯乙烷、四氯化碳、对硝基氯苯、三溴甲烷和1, 2-二溴乙烷等)脱卤转化为低卤代和无卤代化合物, 转化途径主要包括脱卤化氢反应、加氢脱卤反应、脱双卤反应等[24–27]。除了这些低分子量的卤代有机污染物, FeS还能促进高分子量持久性有机污染物(POPs)的非生物转化。Liuet al.[28]报道了FeS能对POPs林丹(γ-六氯环己烷)进行还原脱氯, 在25 ℃、pH 6.9条件下林丹的还原脱氯半衰期为 55 d, 主要通过脱氯化氢和双氯消除等反应逐步脱氯转化为五氯环己烯、四氯环己烯和二氯环己二烯等中间产物以及三氯苯、二氯苯和氯苯等最终产物。此外, 当FeS被固定在真菌Itajahiasp. 产生的高分子聚合物时, 林丹的脱氯率在8 h内达到95%[29]。Pirnieet al.[30]的研究结果也表明FeS能在150 h内将56%的有机氯农药滴滴涕(1,1,1-trichloro-2,2-bis(p-chlorophenyl) ethane, DDT)进行还原脱氯。由此可以推测, FeS对这些卤代污染物在地表环境中的迁移转化会产生重要影响。但迄今为止, 还没有关于FeS还原脱溴HBCD的报道。
本文选用两种不同类型的硫酸盐还原菌(一种是嗜酸性硫酸盐还原菌Desulfosporosinussp., 另一种是嗜中性硫酸盐还原菌Desulfomicrobium baculatum)合成两种FeS矿物, 通过一系列的表征手段研究和比较这两种 FeS的表面特征, 并开展批处理实验, 研究它们还原脱溴 HBCD (包括α-,β-和γ-HBCD 三种主要同分异构体)的动力学过程和途径。本文获得的研究结果将有助于更全面地了解HBCD在地表土壤/沉积环境中的自然衰减过程, 并为HBCD的环境风险评估和污染治理提供重要的理论依据。
1 材料与方法
1.1 实验材料
嗜酸性硫酸盐还原菌Desulfosporosinussp.由中山大学生命科学学院提供, 该菌的培养基(每升)成分为: (NH4)2SO40.45 g、KCl 0.05 g、MgSO4·7H2O 0.5 g、KH2PO40.05 g、Ca(NO3)2·4H2O 0.014 g、酵母提取物 0.2 g, 、油 0.92 g、刃天青钠 1.0 mg、FeSO4·7H2O 0.5 g、抗坏血酸0.02 g、巯基乙醇1 g, pH = 4。嗜中性硫酸盐还原菌Desulfomicrobium baculatum购买于中国普通微生物菌种保藏管理中心, 菌种保存号为1.3467。该菌的培养基(每升)成分为: K2HPO40.5 g、NH4Cl 1.0 g、Na2SO41.0 g、CaCl2·2H2O 0.1 g、MgSO4·7H2O 2.0 g、酵母提取物 1.0 g、DL-乳酸钠2.0 g、刃天青钠 1.0 mg、FeSO4·7H2O 0.5 g、维生素C 0. 2 g、巯基乙酸钠0.1 g, pH = 7。HBCD (纯度>95%)购买自 Tokyo Chemical Industry Co. Ltd。
1.2 FeS的生物合成
按体积比为 10%的接种量将上述两种硫酸盐还原菌菌种分别接种到灭菌过的、相应的新鲜培养基中, 在30 ℃条件下厌氧培养3~7 d直至菌液培养物出现黑色沉淀物即 FeS, 然后将菌液转入厌氧手套箱(99.999% N2)中, 加入少量钼酸钠抑制硫酸盐还原菌活性(以排除其在批处理实验中可能对 HBCD还原脱溴的干扰), 以8000 r/min转速离心分离, 去除上清培养液, 合并FeS, 用去离子水多次清洗FeS,然后将FeS保存于手套箱中用于表征分析和批处理还原脱溴实验。嗜酸性硫酸盐还原菌合成的FeS命名为S-FeS, 嗜中性硫酸盐还原菌合成的FeS命名为Z-FeS。
1.3 批处理还原脱溴实验
HBCD还原脱溴动力学批处理实验采用 8 mL厌氧管为反应瓶。首先在手套箱中将5.88 mL的生物合成FeS悬浮液(去离子水或者缓冲溶液配制, 浓度如下文所述)加入到厌氧管中, 然后加入 120 μL 100 mg/L的 HBCD母液(乙醇配制), 混匀, 塞上内层涂有特氟龙的橡胶塞, 拧紧旋盖, 充分保持密闭,将密封好的厌氧管移出手套箱, 置于恒温振荡箱中反应(转速为 200 r/min以保证异相反应体系下 FeS与HBCD充分接触发生反应, 该振荡过程不影响厌氧管的密闭效果), 温度为30 , ℃避光, 定期(反应开始后的第0 h、6 h、12 h、24 h、36 h和48 h)取出3个平行样对HBCD剩余量进行分析。实验考察了FeS投加浓度(0.3 g/L、0.6 g/L和1.2 g/L)对FeS还原脱溴HBCD程度的影响, FeS悬浮液由Mini-Q去离子水(pH = 6.8)配制, 未调节pH值。此外, 实验还考察了pH值(4、6和8)对FeS还原脱溴HBCD程度的影响, pH值由柠檬酸钠-磷酸氢二钠缓冲液调节,FeS的初始浓度为1.2 g/L。
由于HBCD疏水性强(lgKow> 4.5), 在异相反应体系中容易吸附在 FeS表面, 因此本实验分别对固相中HBCD剩余量(吸附在FeS表面并且未被还原脱溴的部分)和液相中HBCD剩余量进行了萃取。首先,样品以10000 r/min的转速离心10 min, 将固相和液相分开, 萃取固相表面吸附的HBCD的过程如下:萃取剂采用甲醇和甲苯的混合液(体积比为 1/1, 加1 mg/L六溴苯为内标), 每次加2 mL萃取剂, 置于恒温振荡箱中振荡7 min, 转速为200 r/min, 然后以8000 r/min地转速离心10 min, 分离萃取液, 萃取2次后, 合并萃取液, 加入少量经酸洗和甲醇洗过的铜片去硫, 氮吹干萃取液, 加入 4 mL正己烷定容,取出1 mL至1.5 mL的细胞瓶中上机GC-ECD (Gas chromatography with an electron capture detector)进行定量分析; 萃取液相中HBCD的过程如下: 加入2 mL萃取剂正己烷(含1 mg/L内标六溴苯), 置于恒温振荡箱中振荡10 min, 静置, 分离萃取液, 萃取2次后, 合并萃取液, 取出1 mL上机GC-ECD分析。固相萃取时 HBCD 萃取回收率为(101.40±0.99)%,内标六溴苯萃取回收率为(100.5±0.5)%, 基本保证吸附在FeS表面的HBCD完全被萃取出来; 液相萃取时HBCD萃取回收率也达到(99.8±1.9)%, 内标六溴苯萃取回收率为(100.3±0.2)%。预实验研究发现整个反应过程中未在液相中检测到 HBCD (仪器检测限为30 μg/L), 这表明HBCD加入到FeS-H2O体系后, 快速地吸附在FeS表面上, 然后在FeS表面发生还原脱溴反应。因此, 本论文主要研究吸附在矿物表面的HBCD的还原脱溴动力学过程。HBCD的还原脱溴量等于HBCD的初始量减去HBCD在固相上的吸附量, HBCD的转化率计算方法为: HBCD转化率=(HBCD初始量– HBCD固相吸附量)/HBCD初始量。
中间产物鉴定实验采用50 mL厌氧瓶为反应瓶,首先在手套箱中将29.7 mL的生物合成FeS悬浮液(0.6 g/L)加入到厌氧瓶中, 然后加入300 μL 100 mg/L的 HBCD 母液(乙醇配制), 混匀, 塞上内层涂有特氟龙的橡胶塞, 拧紧旋盖, 充分保持密闭, 将厌氧瓶密封好后移出手套箱, 置于恒温振荡箱中反应,转速为200 r/min, 温度为25 , ℃避光, 定期(反应开始后的第0 h、6h、18 h和48 h)取出1个样品对其中的还原脱溴产物进行分析。萃取剂采用甲醇和甲苯的混合液(体积比为 1/1)和正己烷, 样品中的固体采用甲醇和甲苯的混合液萃取, 萃取 2次, 每次使用 20 mL萃取剂, 样品中的液体采用正己烷萃取,萃取2次, 每次使用20 mL萃取剂; 合并所有萃取液, 静置待分层后, 分离上层液(含甲苯、正己烷和少量甲醇), 下层液(含甲醇和水)用正己烷萃取 2次,每次使用20 mL萃取剂, 合并所有上层萃取液。将上层萃取液过无水硫酸钠, 加入适量稀盐酸、甲醇和正己烷清洗过的铜片, 然后将样品进行真空旋蒸至2 mL, 再过复合硅胶氧化铝层析柱洗脱液通过真空旋转蒸发至1 mL, 转移至1.5 mL细胞瓶, 氮吹干,加1 mL正己烷至GC-MS (Gas chromatography-Mass spectrometry)进行分析。
复合硅胶氧化铝柱制备: 使用内径为1 cm、长度为 40 cm 层析柱, 正己烷湿法装柱法, 由下至上分别装入少量脱脂棉、6 cm中性氧化铝、2 cm中性硅胶、8 cm酸性硅胶和2 cm无水硫酸钠。其中脱脂棉、氧化铝和硅胶均经甲醇和二氯甲烷各索氏抽提48 h, 无水硫酸钠经450 ℃焙烧5 h, 洗脱剂采用70 mL二氯甲烷/正己烷(体积比为1/1)混合溶液。
1.4 仪器与分析方法
比表面积的分析采用 BET (Brunauer-Emmett-Teller)法, 使用的仪器是 MicroActive公司 ASAP 2020型比表面积分析仪。SEM-EDS (Scanning Electron Microscope-Energy Disperse Spectroscopy)的测定采用德国ZEISS Merlin型高分辨率热场发射扫描电镜,SE检测器, 加速电压为10 kV。XPS (X-ray photoelectron spectroscopy)分析采用Thermo Fisher K-Alpha 型X射线光电子能谱仪, X 射线源为AlKα线, 全元素扫描能量为100 eV, 单元素扫描能量为30 eV, 分析区域 400 μm, 步长 0.05 eV。XRD (X-Ray Diffraction)测定采用德国Bruker公司的D8 ADVANCE仪器, 靶源采用铜靶, 扫描范围 15°~70°, 扫描速度 2 (°)/min,扫描步长 0.01 (°)/步。
HBCD定量分析采用岛津气相色谱仪(GCECD), 升温程序为: 初始温度为60 ℃, 保持1 min,以20 /min℃的升温速率升至260 , ℃再以5 /min℃的升温速率升至310 , ℃保持10 min。反应过程中α-HBCD、β-HBCD和γ-HBCD同分异构体的测定采用Agilent 1200液相色谱- Agilent 6410电喷雾三重四极杆质谱(LC-ESI-MS/MS), 流动相A为甲醇/水(体积比为 90/10), 流动相 B为乙腈, 流速为0.25 mL/min, 洗脱程序为: 初始A/B为90/10(体积比), 在1 min内下降为60/40, 再在4 min内下降至30/70, 最后在 9 min内上升回到初始的 90/10。HBCD动力学反应中间产物的测定采用岛津气相色谱-质谱联用仪(GC-MS), 柱初始温度为100 , ℃保持1 min, 以10 /min℃的升温速率升至300 , ℃保持10 min; MS采用电子轰击源(EI), 离子源温度为250 ;℃接口温度250 , ℃溶剂延长时间5 min; 离子扫描范围为m/z45~700, 扫描时间为 5.5~31 min。
2 结果和讨论
2.1 生物合成FeS矿物的特性分析

图 1 S-FeS (a)和 Z-FeS (b)的 SEM 图Fig. 1 SEM photomicrographs of FeS formed from acidophilic sulfate-reducing bacteria (S-FeS, a)and neutrophilic sulfate-reducing bacteria (Z-FeS, b)
BET分析的结果表明, S-FeS和Z-FeS的比表面积分别为13.35 m2/g和7.64 m2/g, 明显小于Rickard[31]通过化学法合成的FeS的比表面积(36.5 m2/g)。图1是 S-FeS和 Z-FeS的扫描电镜图像, 由图可看出两种硫酸盐还原菌合成的FeS结构均为不定形的蓬松状。图2是两种FeS的X-射线衍射图谱, 可以看出它们仅有几个宽化的峰与四方硫铁矿(PDF15-0037)相吻合, 表明合成的 FeS结晶性较差。柯杭等[32]发现利用脱硫肠状菌(Desulfotomaculum putei)合成的FeS主要以无定形态和微晶形式存在, 而谢翼飞等[33]报道了由脱硫弧菌(Desulfovibriosp.)、脱硫肠状菌(Desulfotomaculumsp.)、脱硫杆菌(Desulfobactersp.)、阴沟肠杆菌(Enterobacter cloacesp.)、芽孢杆菌(Bacillussp.)等5株菌组成的复合菌合成的FeS也主要以无定形态和微晶形式存在。另外, Rickard[34]研究表明, FeS在25 ℃的水溶液中转化成结晶较好的四方硫铁矿需要两年时间; 这也间接说明硫酸盐还原菌初期合成的FeS结晶较差是合理的。

图2 S-FeS和Z-FeS的XRD谱图以及四方硫铁矿的标准XRD谱图Fig. 2 XRD patterns of S-FeS and Z-FeS samples as well as the standard mackinawite
SEM-EDS测出S-FeS和Z-FeS样品的铁硫原子比(Fe/S)分别为 0.91和 0.84。由此可以看出本文所合成的两种FeS矿物并不是由单一的FeS组成。这与柯杭等[32]的研究结果相吻合, 他们发现硫酸盐还原菌Desulfotomaculum putei合成的FeS矿物的Fe/S也不是11∶。事实上, 已有研究发现, 在自然界的沉积物中发现的铁硫化物矿物主要有无定形 FeS、四方硫铁矿、硫复铁矿和黄铁矿[23]; 其中, 无定形FeS、四方硫铁矿和硫复铁矿在酸性条件下易分解,常转化为热化学更稳定的黄铁矿, 但是在没有过量HS–存在的条件下, 它们也可长期存在。不过Lennieet al.[35]和 Pósfaiet al.等[36]的研究认为, 硫酸盐还原菌与 Fe2+形成的硫铁矿初期主要是结晶较差的四方硫铁矿。Benninget al.[37]和Wilkinet al.[38]的研究也表明, 处于初生态的硫铁化合物主要以无定形 FeS和四方硫铁矿为主。如图2所示, S-FeS的XRD衍射图有 4 个衍射峰(2θ= 17.6°、30.1°、39.0°和 50.4°)分别对应四方硫铁矿(15-0037)的(001)、(101)、(111)和(200)面网衍射峰, Z-FeS有2个衍射峰(2θ= 17.6°和 50.4°)分别对应四方硫铁矿的(001)和(200)面网衍射峰; 因此可初步推断S-FeS和Z-FeS均是主要由初生成的无定形FeS和结晶较差的四方硫铁矿组成。
图3为S-FeS和Z-FeS的O(1s)、Fe(2p3/2)和S(2p)的XPS谱图, 并进行了分峰处理。表1列出了S-FeS和Z-FeS的O(1s)、Fe(2p3/2)和S(2p)谱峰拟合出来的相应化合物及相关参数。根据拟合的化合物数据并参考文献[39–45], 笔者推论两种 FeS样品的 O(1s)峰的结合能(530.0±0.1) eV、(531.3±0.1) eV 和(532.4±0.1) eV分别对应铁氧化合物的氧[39]、羟基氧化物的氧和吸附水的氧[40]。从表 1中可以看出, 两种 FeS矿物表面羟基氧化物的含量几乎相同, 但它们表面的铁氧化物和吸附水含量差异较大。Fe(2p3/2)峰的结合能(707.3±0.2) eV、(708.2±0.3) eV 和(709.5±0.2) eV对应与硫结合的二价铁(Fe(II)-S)[40–41], 它在 S-FeS和Z-FeS的相对含量分别为63.6%和77.2%, 这与笔者之前的推论是吻合的, 也就是说两种FeS的主要组成成分都是四方硫铁矿和 FeS; Fe(2p3/2)峰的结合能(711.0±0.2) eV对应的化合物为与硫结合的三价铁(Fe(III)-S)[41], 结合能(711.8±0.4) eV 和(714.3±0.1) eV对应的化合物为与氧结合的三价铁(Fe(Ⅲ)-O)[42–43]。S(2p)峰的结合能(161.1±0.2) eV 和(162.3±0.2) eV 对应 S2–[39,44], S2–在-FeS和 Z-FeS的相对含量分别为67.7%和 76.5%, 与 Fe(Ⅱ)的相对含量非常接近, 表明大部分S2–与Fe结合而形成FeS; S(2p)峰的结合能(163.8±0.2) eV、(167.2±0.4) eV 和(168.4±0.1) eV 分别对应的是 Sn2–[44]、SO32–和 SO42–[39,44]。SO32–和SO42–相对含量均小于4.5%, 可忽略不计。

表1 两种FeS的S(2p)、Fe(2p3/2)和O(1s)XPS谱峰的结合能和峰面积Table 1 XPS binding energies (BE) and relative abundances of O(1s), Fe(2p3/2) and S(2p) on the surface of S-FeS and Z-FeS
2.2 生物合成FeS还原脱溴HBCD的反应动力学
如图4所示, S-FeS和Z-FeS均对HBCD具有还原脱溴的能力, 而且还原脱溴反应都基本遵循假一级反应动力学, 其相关参数如表2所示。已有研究表明, 还原型多硫化物和纳米铁对HBCD的还原脱溴也都符合假一级反应动力学[21,39]。在本研究中, 当 FeS起始浓度在0.3~1.2 g/L范围内变化时, 随着FeS浓度的增高,FeS还原脱溴HBCD的速率常数也是呈增加趋势, 并且当FeS的浓度为1.2 g/L时, 两种FeS对HBCD的还原脱溴率在12小时内均达到100%。这现象可能与高浓度的FeS能提供更多的活性反应位点有关。类似地,罗丽卉等[45]在研究由硫酸盐还原菌Desulfovibrio desulfuricans合成FeS的时候发现, 该FeS材料去除Cu2+的效率与其起始浓度呈正相关关系。
通常认为非生物还原剂的比表面积越大其反应活性越高[39]。如表2所示, 当FeS的起始浓度为0.3 g/L和0.6 g/L时, S-FeS还原脱溴HBCD的速率常数与Z-FeS非常接近, 尽管它的比表面积大于Z-FeS; 这很可能是因为Z-FeS表面活性成分FeS的含量高于S-FeS表面FeS的含量(表1), 抵消了S-FeS由于具有更大比表面积而带来的活性优势。当FeS的起始浓度达到1.2 g/L时, S-FeS还原脱溴HBCD的速率常数甚至略小于 Z-FeS; 这很可能是因为随着起始浓度的进一步增加, S-FeS颗粒的聚合程度会有所提高[39], 削弱了其在比表面积方面的优势, 而 Z-FeS在表面活性成分FeS含量方面的优势由于起始浓度提高而得到加强。

图 3 S-FeS (a、b和 c)和 Z-FeS (d、e和 f)的 O(1s)、Fe(2p3/2)和 S(2p)的 XPS分峰谱图Fig. 3 XPS spectra of O(1s), Fe(2p3/2) and S(2p) in S-FeS and Z-FeS samples, respectively
实验考察了反应溶液pH值(4、6、8)对FeS还原脱溴HBCD效果的影响; 结果如图5所示。pH变化对两种FeS还原脱溴HBCD的影响基本相同, 即HBCD的还原脱溴率随着 pH值的增加而提高。当pH值为4时, HBCD基本没有发生还原脱溴, 这可能与酸性条件下FeS表面活性组分发生溶解有关[46];当pH值为6时, 48 h内S-FeS和Z-FeS对HBCD的还原脱溴率分别为50.8%和65.7%; 当pH值提高至8时, 48 h内S-FeS和Z-FeS对HBCD的还原脱溴率提高至 93.4%和 90.9%。Butler[46–47]和 Choi等[48]的研究也发现FeS对氯代溶剂(如六氯乙烷、三氯乙烯和三氯乙烷)的还原脱溴率随着 pH值的增加而提高。pH值升高能提高FeS对HBCD的还原脱溴率可能是由以下两方面的原因造成的[47–48]: (1) pH值升高能促进 FeS表面去质子化活性基团的形成, 而这些活性基团既能作为电子载体促进电子转移至卤代污染物, 也能作为亲核基团诱导卤代污染物的还原双脱卤过程; (2) FeS或者四方硫铁矿在低pH值条件下不稳定, 容易氧化形成硫复铁矿, 降低还原脱卤活性。值得注意的是同一 pH值条件下 S-FeS和Z-FeS对 HBCD的还原脱溴率差异不大, 说明这两种不同硫酸盐还原菌合成的FeS的还原脱卤活性差异不大。另外, 未加缓冲液调节的反应体系(初始溶液pH = 6.8, 1.2 g/L FeS)下HBCD在12 h内的还原脱溴就率达到 100%, 然而添加了柠檬酸钠-磷酸氢二钠缓冲液调节的反应体系(pH = 6或者8, 1.2 g/L FeS)下HBCD在48 h内的还原脱溴率不到94%, 这可能和缓冲液与FeS表面的铁反应生成磷酸盐沉淀引起矿物表面钝化从而降低反应速率有关[49]。
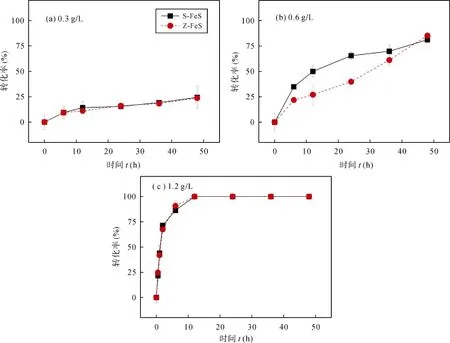
图4 在不同FeS起始浓度条件下HBCD的还原脱溴速率随时间变化图Fig. 4 The reductive debromination percentage (%) of HBCD by S-FeS and Z-FeS at different initial FeS concentrations of 0.3 g/L (a),0.6 g/L (b) and 1.2 g/L (c), respectively.
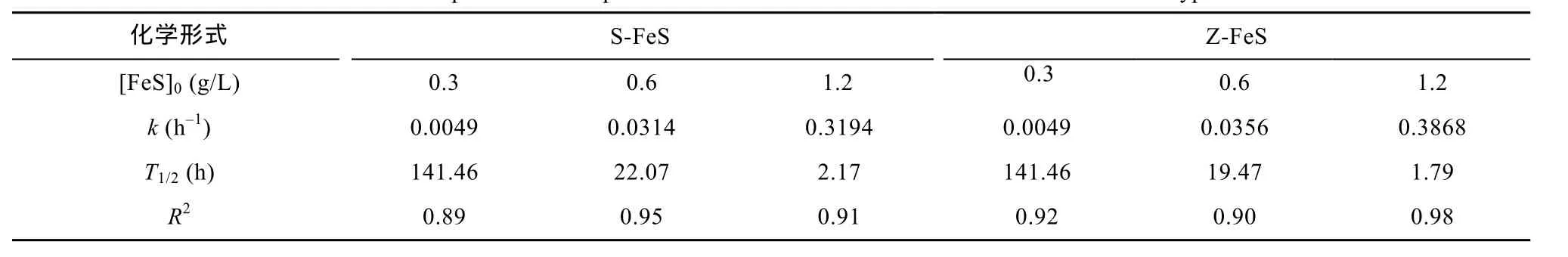
表2 两种FeS还原脱溴HBCD的假一级反应速率常数及半衰期Table 2 The fitted parameters for pseudo first-order reactions between HBCD and the two types of FeS
由图 6 可知, S-FeS 和 Z-FeS 对α-、β-和γ-HBCD等三种主要的 HBCD异构体都具有还原脱溴能力,还原脱溴速率随着 FeS起始浓度的增加而增加; 并且它们对这三种异构体的还原脱溴速率大小顺序均依次为β-HBCD >γ-HBCD >α-HBCD。这与 Lo 等报道的在厌氧条件下还原型多硫化物对γ-HBCD的还原脱溴速率显著高于α-HBCD的结果是吻合的[22]。这些发现说明在某些条件下HBCD的非生物转化具有一定的同分异构选择性,β-HBCD和γ-HBCD更倾向于被优先还原脱溴; 这也间接解释了为什么在一些受HBCD污染的沉积物与土壤中α-HBCD在三种主要异构体中所占的比例高于其在工业生产的HBCD中的比例, 甚至高于γ-HBCD所占的比例[50]。
2.3 反应机理研究

图5 不同pH条件下FeS对HBCD的还原脱溴率随时间变化Fig.5 The reductive debromination percentage (%) of HBCD by S-FeS and Z-FeS at different pH adjusted by citric acid-disodium hydrogen phosphate buffer
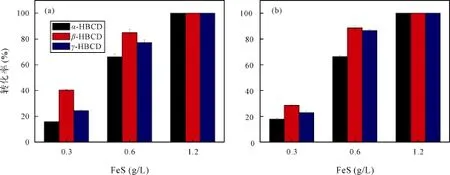
图 6 反应48 h 后S-FeS (a)和Z-FeS (b)体系下 α-、β-和γ-HBCD的还原脱溴率Fig.6 The reductive debromination percentage (%) of α, β-, γ-HBCD in S-FeS (a) and Z-FeS (b) system after 48-h reaction
本研究发现在HBCD与S-FeS或者Z-FeS的反应过程中, GC-MS都检测到了三个还原脱溴产物峰,而且产物生成变化趋势基本相同。下文以 S-FeS为例展示了HBCD转化过程中三个还原脱溴产物的生成和变化(图7)。通过和标准品或者文献报道的还原脱溴产物质谱图的碎片质量、相对丰度以及母体化合物的结构比对, 鉴定出三种中间产物分别为四溴环十二碳烯(TBCD, C12H18Br4)、二溴环十二碳二烯(DBCDD, C12H18Br2)和环十二碳三烯(CDT, C12H18)。这些还原脱溴产物的主要碎片离子峰和相对分子量见表3。TBCD和 DBCDD的鉴定主要通过和文献[51–52]报道的TBCD和DBCDD的质谱图比对, CDT的鉴定通过和标准品的质谱图比对。据了解, 目前仅有两篇文献报道了HBCD非生物还原脱溴的中间产物:Loet al.[22]发现在厌氧条件下还原型多硫化物还原脱溴 HBCD生成两种中间产物, 可能是 TBCD和DBCDD, 但没有检测到完全脱溴的产物 CDT; 而Tsoet al.[53]报道了在缺氧条件下纳米铁能还原脱溴HBCD生成两种中间产物, 分别是DBCDD和CDT。另外, Daviset al.[16]在研究废水污泥和淡水沉积物中 HBCD的微生物还原脱溴时, 发现了三种中间产物, 分别是TBCD、DBCDD和CDT。
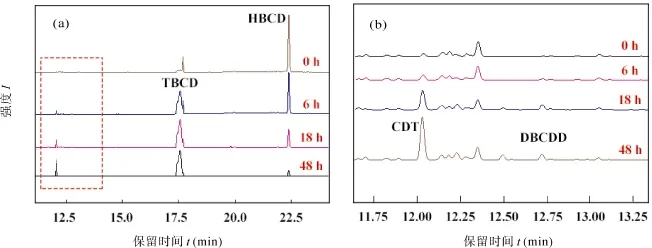
图7 HBCD的还原脱溴产物随时间变化的色谱图 ((b) 是放大以后的 (a) 中用虚线标出的部分)Fig.7 The chromatograms and dynamics of reductive debromination products of HBCD by S-FeS((b) an enlarged section of (a) marked by dotted lines)

表3 HBCD及其还原脱溴中间产物的质谱鉴定表Table 3 Mass spectra data for HBCD and its reductive debromination products by FeS

图8 FeS还原脱溴HBCD的反应途径图Fig.8 The proposed reductive debromination pathway of HBCD by FeS
随着HBCD还原脱溴的进行, 三种产物生成的相对含量呈现不同的变化趋势(图7)。反应初始(0 h),检测到少量TBCD和CDT, 这可能与气相色谱分析时HBCD在高温条件下会热解有关[51]。HBCD发生还原脱溴反应6 h后, TBCD的量显著增加, 而其他产物无明显增加, 表明反应初始阶段HBCD主要还原脱溴为TBCD。HBCD与FeS反应18 h后, TBCD的量持续积累, CDT的量也明显增加, 并且检测到少量 DBCDD, 这表明 HBCD进一步脱溴转化为低溴代和无溴代产物。反应48 h后, TBCD的积累量降低, DBCDD量增加不显著, 而CDT的量持续积累,由此推测CDT是反应的终产物, DBCDD积累量低可能与其不稳定容易发生脱溴转化有关。据文献报道, FeS还原脱卤的机理包括脱卤化氢反应、加氢脱卤反应和脱双卤反应等[24–27]。不过本研究未检测到五溴代中间产物的生成, 基本排除HBCD发生脱溴化氢和加氢脱溴反应生成五溴环十二碳烯和五溴环十二烷的可能性。根据三种中间产物的结构以及相对含量的变化趋势, 可以推测 S-FeS或 Z-FeS还原脱溴 HBCD的途径可能以邻位双脱溴为主(图 8):HBCD被FeS还原, 先脱掉两个Br, 生成TBCD, 然后TBCD再继续被还原, 脱掉两个Br, 生成DBCDD,接着DBCDD被进一步还原, 再掉两个Br生成不含Br的中间产物CDT, 从而降低了HBCD的毒性。关于硫铁矿物参与卤代污染物的还原脱溴的反应机理解释有几种[46,54]: (1) 溶液中的亚铁离子和FeS矿物表面的二价态铁被氧化形成三价态的铁与卤代化合物形成一个自催化的氧化还原系统, 或者有机络合态亚铁离子可直接参与还原脱卤反应; (2) 硫铁矿物表面结合态的亚铁可充当电子供体使得卤代化合物还原脱卤; (3) 硫铁矿物表面的结合态硫是真正的活性组分, 具有还原脱卤作用。笔者前期研究发现溶解态亚铁离子和HBCD不发生反应或者添加亚铁离子并不促进FeS对HBCD的还原脱溴速率, 基本否定了机理解释 (1) 和 (2) 存在的可能性。另外,Loet al.[22]的研究证明了溶解态硫离子作为亲核基团可通过一步反应, 同时失去两个电子, 促进HBCD的邻位双脱溴反应生成两种中间产物 TBCD和 DBCDD, 表明硫铁矿物表面结合态硫有可能参与HBCD的还原脱溴反应。为了深入了解反应机理,后续研究需要进一步对矿物表面的形态结构变化、矿物颗粒与 HBCD的结合状态, 以及中间过渡体的类型结构进行分析。总体来说, FeS对HBCD的还原脱溴生成TBCD、DBCDD和CDT是迄今为止文献报道的关于 HBCD非生物还原脱溴的最为‘完整’的还原脱溴途径, 尽管Daviset al.[13]在2006年就已经发现: 废水污泥和淡水沉积物中的微生物可以通过上述途径将HBCD还原脱溴为CDT。但必须指出的是, 目前学界并不了解CDT能否被非生物还原剂进一步还原转化。因此, 这个问题应该在未来的研究中得到更多关注。此外, 应该定量化地研究生物合成的FeS矿物在各种真实环境下HBCD的自然衰减过程中的作用。
3 结 论
(1) S-FeS和Z-FeS的主要组分均为初生成的无定形FeS和结晶较差的四方硫铁矿;
(2) S-FeS具有较大的比表面积, 但其表面的活性组分FeS的含量低于Z-FeS;
(3) S-FeS和Z-FeS对HBCD的还原脱溴都遵循假一级动力学; 速率常数呈现随FeS起始浓度(范围0.3~1.2 g/L)增大而增大的趋势;
(4) 反应溶液 pH值(4~8)的升高能促进 FeS对HBCD的还原脱溴率;
(5) S-FeS和Z-FeS都能还原脱溴HBCD的三种主要异构体, 还原脱溴率大小顺序都依次为β-HBCD>γ-HBCD >α-HBCD;
(6) S-FeS和Z-FeS都能将HBCD还原脱溴生成TBCD、DBCDD和CDT。
:
[1] UNEP. Additional information on hexabromocyclododecane(HBCD) for Stockholm Convention [C]. Persistent Organic Pollutants Review Committee, UNEP, 2013.
[2] Law R J, Allchin C R, de Boer J, Covaci A, Herzke D, Lepom P, Morris S, Tronczynski J, de Wit C A. Levels and trends of brominated flame retardants in the European environment [J].Chemosphere, 2006, 64(2): 187–208.
[3] Tomy G T, Budakowski W, Halldorson T, Whittle DM, Keir MJ, Marvin C, MacInnis G, Alaee M. Biomagnification of αand γ- hexabromocyclododecane isomers in a Lake Ontario food web [J]. Environ Sci Technol, 2004, 38(8): 2298–2303.
[4] Remberger M, Sternbeck J, Palm A, Kaj L, Stromberg K,Brorstrom-Lunden E. The environmental occurrence of hexabromocyclododecane in Sweden [J]. Chemosphere, 2004,54(1): 9–21.
[5] Eljarrat E, de la Cal A, Raldua D, Duran C, Barceló D. Occurrence and bioavailability of polybrominated diphenyl ethers and hexabromocyclododecane in sediment and fish from the Cinca River, a tributary of the Ebro River (Spain) [J].Environ Sci Technol, 2004, 38(9): 2603–2608.
[6] Marvin C H, Tomy G T, Alaee M, Macinnis G. Distribution of hexabromocyclododecane in Detroit River suspended sediments [J]. Chemosphere, 2006, 64(2): 268–275.
[7] Stapleton H M, Dodder N G, Kucklick J R, Reddy C M,Schantz M M, Becker P R, Gulland F, Porter B J, Wise S A.Determination of HBCD, PBDEs and MeO-BDEs in California sea lions (Zalophus californianus) stranded between 1993 and 2003 [J]. Mar Pollut Bull, 2006, 52(5): 522–531.
[8] Verreault J, Gabrielsen G V, Chu S G, Muir D, Andersen M,Hamaed A, Letcher R J. Flame retardants and methoxylated and hydroxylated polybrominated diphenyl ethers in two Norwegian Arctic top predators: Glaucous gulls and polar bears [J]. Environ Sci Technol, 2005, 39(16): 6021–6028.
[9] de Wit C A, Alaee M, Muir D C G. Levels and trends of brominated flame retardants in the Arctic [J]. Chemosphere, 2006,64(2): 209–233.
[10] Goscinny S, Vandevijvere S, Maleki M, van Overmeire I,Windal I, Hanot V, Blaude M, Vleminckx C, van Loco J. Dietary intake of hexabromocyclododecane diastereoisomers(alpha-, beta-, and gamma-HBCD) in the Belgian adult population [J]. Chemosphere, 2011, 84(3): 279–288.
[11] Zhang X, Yang F, Xu C, Liu W P, Wen S, Xu Y. Cytotoxicity evaluation of three pairs of hexabromocyclododecane (HBCD)enantiomers on Hep G2 cell [J]. Toxicol In Vitro, 2008, 22(6):1520–1527.
[12] Zhang J, Williams T D, Abdallah M A, Harrad S, Chipman, J K, Viant, M R. Transcriptomic and metabolomic approaches to investigate the molecular responses of human cell lines exposed to the flame retardant hexabromocyclododecane(HBCD) [J]. Toxicol In Vitro, 2015, 29(8): 2116–2123.
[13] Davis J W, Gonsior S J, Markham D A, Friederich U, Hunziker R W, Ariano J M. Biodegradation and product identification of [C-14] hexabromocyclododecane in wastewater sludge and freshwater aquatic sediment [J]. Environ Sci Technol, 2006, 40(17): 5395–5401.
[14] Heeb N V, Zindel D, Geueke B, Kohler H E, Lienemann P.Biotransformation of hexabromocyclododecanes (HBCDs)with LinB-an HCH-converting bacterial enzyme [J]. Environ Sci Technol, 2012, 46(12): 6566–6574.
[15] Yamada T, Takahama Y, Yamada Y. Isolation ofPseudomonassp. strain HB01 which degrades the persistent brominated flame retardant gamma-hexabromocyclododecane [J]. Biosci Biotechnol, Biochem, 2009, 73(7): 1674–1678.
[16] Davis J W, Gonsior S, Marty G, Ariano J. The transformation of hexabromocyclododecane in aerobic and anaerobic soils and aquatic sediments [J]. Water Res, 2005, 39(6): 1075–1084.
[17] Gerecke A C, Giger W, Hartmann P C, Heeb N V, Kohler H E,Schmid P, Zennegg M, Kohler M. Anaerobic degradation of brominated flame retardants in sewage sludge [J]. Chemosphere, 2006, 64(2): 311–317.
[18] Zhao Y, Zhang X, Sojinu O S S. Thermodynamics and photochemical properties of alpha, beta, and gamma-hexabromocyclododecanes: A theoretical study [J]. Chemosphere, 2010,80(2): 150–156.
[19] Harrad S, Abdallah M A, Covaci A. Causes of variability in concentrations and diastereomer patterns of hexabromocyclododecanes in indoor dust [J]. Environ Int, 2009, 35(3):573–579.
[20] 高亚杰, 张娴, 颜昌宙. 水环境中六溴环十二烷的光降解研究[J]. 环境化学, 2011, 30(3): 598–603.Gao Ya-jie, Zhang Xian, Yan Chang-zhou. Photodegradation of hexabromocyclododecanes in water [J]. Environ Chem,2011, 30(3): 598–603 (in Chinese with English abstract).
[21] Covaci A, Gerecke A C, Law R J, Voorspoels S, Kohler M,Heeb N V, Leslie H, Allchin C R, de Boer J. Hexabromocyclododecanes (HBCDs) in the environment and humans: A review [J]. Environ Sci Technol, 2006, 40(12): 3679–3688.
[22] Lo K W, Saha-Roy S C, Jans U. Investigation of the reaction of hexabromocyclododecane with polysulfide and bisulfide in methanol/water solutions [J]. Chemosphere, 2012, 87(2): 158–162.
[23] Ward J C. The structure and properties of some iron sulphides [J].Rev Pure Appl Chem, 1970, 20: 175–206.
[24] Butler E C, Hayes K F. Kinetics of the transformation of trichloroethylene and tetrachloroethylene by iron sulfide [J].Environ Sci Technol, 1999, 33(12): 2021–2027.
[25] Butler E C, Hayes K F. Kinetics of the transformation of halogenated aliphatic compounds by iron sulfide [J]. Environ Sci Technol 2000, 34(3): 422–429.
[26] Elsner M, Schwarzenbach R P, Haderlein S B. Reactivity of Fe(II)-bearing minerals toward reductive transformation of organic contaminants [J]. Environ Sci Technol, 2004, 38(3):799–807.
[27] Kuder T, Wilson J T, Philp P, He Y T. Carbon isotope fractionation in reactions of 1, 2-dibromoethane with FeS and hydrogen sulfide [J]. Environ Sci Technol, 2012, 46(14):7495–7502.
[28] Liu X M, Peng P A, Fu J M, Huang W L. Effects of FeS on the transformation kinetics of gamma-hexachlorocyclohexane [J].Environ Sci Technol, 2003, 37(9): 1822–1828.
[29] Paknikar K M, Nagpal V, Pethkar A V, Rajwade J M. Degradation of lindane from aqueous solutions using iron sulfide nanoparticles stabilized by biopolymers [J]. Sci Technol Adv Mat, 2005, 6(s3/4): 370–374.
[30] Pirnie E F, Talley J W, Hundal L S. Abiotic transformation of DDT in aqueous solutions [J]. Chemosphere, 2006, 65(9):1576–1582.
[31] Rickard D. Kinetics of pyrite formation by the H2S oxidation of iron (II) monosulfide in aqueous solutions between 25 and 125℃: The rate equation [J]. Geochim Cosmochim Acta, 1997,61(1): 115–134.
[32] 柯杭, 李莉, 张旭. 生物合成硫化亚铁对地下水中六价铬的去除效果研究[C]. 中国环境科学学会学术年会浦华环保优秀论文集. 昆明: 中国环境科学学会, 2013: 174–179.Ke Hang, Li Li, Zhang Xu. Effect of biological iron sulfide on Cr(VI)-containing groundwater treatment [C]. Proceedings of Annual Conference of Chinese Society for Environmental Sciences on Puhua Environmental Protection. Kunming: Chinese Society for Environmental Sciences, 2013: 174–179 (in Chinese).
[33] 谢翼飞, 李旭东, 李福德. 生物硫铁复合材料处理含铬废水及铬资源化研究[J]. 中国环境科学, 2009, 29(12): 1260–1265.Xie Yi-fei, Li Xu-dong, Li Fu-de. Application of biological iron sulfide composites in chromium-containing wastewater treatment and chromium reclamation [J]. China Environ Sci,2009, 29(12): 1260–1265 (in Chinese with English abstract).
[34] Rickard D. Kinetics of FeS precipitation: Part 1. Competing reaction mechanisms [J]. Geochim Cosmochim Acta, 1995,59(21): 4367–4379.
[35] Lennie A R, Redfern S A, Champness P E, Stoddart C P,Schofield P F, Vaughan D J. Transformation of mackinawite to greigite: An in situ X-ray powder diffraction and transmission electron microscope study [J]. Am Mineral, 1997, 82(3):302–309.
[36] Pósfai M, Buseck P R, Bazylinski D A, Frankel R B. Iron sulfides from magnetotactic bacteria: Structure, composition, and phase transitions [J]. Am Mineral, 1998, 83(11): 1469–1481.
[37] Benning L G, Wilkin R T, Barnes H L. Reaction pathways in the Fe-S system below 100 degrees [J]. Chem Geol, 2000,167(1/2): 25–51.
[38] Wilkin R T, Barnes H L. Pyrite formation by reactions of iron monosulfides with dissolved inorganic and organic sulfur species [J]. Geochim Cosmochim Acta, 1996, 60(21): 4167–4179.
[39] Mandrino D. XPS and SEM of unpolished and polished FeS surface [J]. Mater Sci Technol, 2011, 45(4): 325-328.
[40] Herbert R B, Benner S G, Pratt A R, Blowes D W. Surface chemistry and morphology of poorly crystalline iron sulfides precipitated in media containing sulfate-reducing bacteria [J].Chem Geol, 1998, 144(1): 87–97.
[41] Liang C H, Wang H, Huang N B. Effects of sulphate-reducing bacteria on corrosion behaviour of 2205 duplex stainless steel [J].J Iron Steel Res Int, 2014, 21(4): 444–450.
[42] Mcintyre N S, Zetaruk D G. X-ray photoelectron spectroscopic studies of iron oxides[J]. Anal. Chem, 1977, 49(11):1521–1529.
[43] Jeong H Y, Han Y S, Park S W, Hayes K F. Aerobic oxidation of mackinawite (FeS) and its environmental implication for arsenic mobilization [J]. Geochim Cosmochim Acta, 2010,74(11): 3182–3198.
[44] Neal A L, Techkarnjanaruk S, Dohnalkova A, Mccready D,Peyton B M, Geesey G G. Iron sulfides and sulfur species produced at hematite surfaces in the presence of sulfate-reducing bacteria [J]. Geochim Cosmochim Acta, 2001,65(2): 223–235.
[45] 罗丽卉, 谢翼飞, 李旭东. 生物硫铁复合材料处理含铜废水及机理研究[J]. 中国环境科学, 2012, 32(2): 249–253.
Luo Li-hui, Xie Yi-fei, Li Xu-dong. Biological iron sulfide composites in the treatment of copper-contaminated wastewater and its mechanism [J]. China Environ Sci, 2012, 32(2):249–253 (in Chinese with English abstract).
[46] Butler E C, Hayes K. Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide [J]. Environ Sci Technol, 1998, 32(9): 1276–1284.
[47] Butler E C, Hayes K. Factors influencing rates and products in the transformation of trichloroethylene by iron sulfide and iron metal [J]. Environ Sci Technol, 2001, 35(19): 3884–3891.
[48] Choi J, Choi K, Lee W. Effects of transition metal and sulfide on the reductive dechlorination of carbon tetrachloride and 1,1, 1-trichloroethane by FeS [J]. J Hazard Mater, 2009, 162(2):1151–1158.
[49] Kim E J, Murugesan K, Kim J H, Tratnyek P G, Chang Y S.Remediation of trichloroethylene by FeS-coated iron nanoparticles in simulated and real groundwater: Effects of water chemistry [J]. Ind Eng Chem Res, 2013, 52(27): 9343–9350.
[50] 张艳伟. 六溴环十二烷异构体及其对映体的环境分布与生物富集[D]. 天津: 南开大学, 2014.Zhang Yan-wei. Environmental distribution and bioaccumulation of hexabromocyclododecane diastereomers and enantiomers [D].Tianjin: Nankai University, 2014 (in Chinese with English abstract).
[51] Barontini F, Cozzani V, Cuzzola A, Petarca L. Investigation of hexabromocyclododecane thermal degradation pathways by gas chromatography/mass spectrometry [J]. Rapid Commun Mass Spectrom, 2001, 15(9): 690–698.
[52] Davis J W, Gonsior S J, Markham D A, Friederich U, Hunziker R W, Ariano J M. Biodegradation and product identification of [14C] hexabromocyclododecane in wastewater sludge and freshwater aquatic sediment [J]. Environ Sci Technol,2006, 40(17): 5395–5401.
[53] Tso C P, Shih Y H. The transformation of hexabromocyclododecane using zerovalent iron nanoparticle aggregates [J]. J Hazard Mater, 2014, 277(4): 76–83.
[54] 刘相梅. 硫化亚铁体系下林丹的非生物转化[D]. 北京: 中国科学院研究生院, 2002.Liu Xiang-mei. Abiotic transformation of γ-hexachlorocyclohexane(HCH) in FeS system [D]. Beijing: Graduate University of Chinese Academy of Sciences, 2002 (in Chinese with English abstract).
