杨梅S-SAP分子标记体系的建立及应用
2016-06-21舒巧云刘珠琴章建红
焦 云,舒巧云,刘珠琴,章建红
(宁波市农业科学研究院 林业研究所,浙江 宁波 315040)
杨梅S-SAP分子标记体系的建立及应用
焦 云,舒巧云,刘珠琴,章建红
(宁波市农业科学研究院 林业研究所,浙江 宁波 315040)
摘要:基于杨梅全基因组Ty1-copia类型逆转座子LTR序列设计开发S-SAP引物组合,利用48份杨梅试材对其多态性指数进行评价,使用UPGMA(Unweighted Pair Group Method with Arithmatic Mean)法,构建了系统聚类树,并进行了二维和三维主坐标分析,进而探讨了杨梅种质资源遗传亲缘关系。成功开发了18个杨梅S-SAP引物组合,共扩增获得3739个条带,平均每个引物组合可扩增208个条带(100~500 bp),每个引物组合扩增条带数量范围为98~292,Shannon信息指数范围为0.2041~0.4522。其中,引物组合JY20的多态性最高,可扩增出292个条带。基于18个S-SAP引物组合,使用UPGMA法构建所有试材的系统聚类树,结果共分为3个组及3个亚组。聚类分析与主坐标分析结果高度相似,具有相同地域来源的品种大部分被划分于同一个分组,使用聚类分析与主坐标分析所得结果基本一致,但是聚类结果与果实色泽没有明显的关联性,利用三维主坐标分析更能全面且真实地反映杨梅品种间的亲缘关系。
关键词:杨梅;S-SAP;分子标记;遗传多样性
随着分子生物学技术的发展,现在DNA分子标记技术已有数十种,广泛应用于遗传育种、基因组作图、基因定位、物种亲缘关系鉴别、基因库构建、基因克隆等方面。其中,逆转座子分子标记之一的S-SAP(Sequence-Specific Amplification Polymorphism)是根据AFLP演变而来,其扩增的多态性一方面来源于限制性位点及其两侧序列的变异,与AFLP检测的变异相同;另一方面来源于LTR 5′末端的变异和反转录转座子插入位点的变异,在理论上其多态性高于AFLP。与其他类型分子标记相比,S-SAP分子标记的多态性较高,利用其评价获得的植物分类及进化分析结果更接近于基于地理和形态上的分析结果,进而可以非常有效地进行品种或基因型的鉴定,尤其在芽变性状的鉴定方面更具灵敏性[1-2],因此,它被认为是多态性最丰富、灵敏度最高、反映的多态信息含量最多的一种分子标记类型。
目前,S-SAP分子标记技术已经成功应用于葡萄[1]、苹果[2]、桃[3]等果树的遗传多样性分析,由于逆转座子引物的开发具有种族特异性,不同物种之间的逆转座子引物异质性很大,至今尚无S-SAP分子标记技术在杨梅遗传多样性分析应用的报道。本研究基于前期杨梅全基因组测序结果,深入挖掘Ty1-copia类逆转座子长末端重复序列LTR(Long Terminal Repeat)信息,并建立适合于杨梅的S-SAP分子标记技术体系,为进一步利用该标记开展杨梅遗传多样性分析及相关基因组学研究奠定基础。
1材料与方法
1.1材料
从不同省份收集48份杨梅材料用于S-SAP分子标记开发及遗传多样性分析,具体来源见表1。从健康植株上分别采集无病虫害的嫩叶,放入预先准备好的液氮中迅速冷冻,然后转入-80 ℃冰箱中保存,杨梅基因组DNA使用快速离心柱型植物基因组DNA提取试剂盒(目录号:DP3112,百泰克)进行提取。

表1 48份实验材料相关信息
1.2实验方法
1.2.1S-SAP分子标记引物设计使用逆转座子预测软件LTR_STRUC version 1.1对杨梅全基因组DNA序列进行分析。在预测结果数据中优先选择两端LTR序列相似度大于99%的逆转座子,使用Primer Premier 5.0并参考3′端LTR区域序列,设计S-SAP分子标记中选择性扩增所需的下游引物,在设计该引物时仅选择从本区域5′端第一个碱基开始,同时替换第一个碱基为错配碱基,设计引物长度为19~23 bp,在特异选择性扩增引物末端添加4个选择性碱基(GAG、CGT、GGT、TCC),从而形成不同引物组合,同时在其5′端添加18 bp的通用引物序列(M13)TGTAAAACGACGGCCAGT,Tail中的5′端分别添加3种不同的荧光基团,即FAM、HEX和PET,用于后期使用遗传分析仪进行扩增产物的检测与分析。相关引物序列见表2,委托上海英骏生物技术有限公司合成,用超纯水溶解。
1.2.2双酶切、预扩增及选择性扩增具体方法参照相关文献[3]进行。
1.2.3基因型分型检测与数据分析取上述选择性扩增产物等比例混合后,委托上海美吉生物医药科技有限公司在ABI 3730遗传分析仪上进行检测。扩增产物片段大小用Genemapper 4.0(Applied Biosystems, Foster City, CA, USA)软件进行读数,只统计长度范围在100~500 bp内的扩增产物片段,然后使用Microsoft Excel 2007构建数据矩阵表(1表示有条带,0表示无条带);使用POPGENE Version 1.32软件计算所有引物组合的Shannon信息指数和有效等位基因数;基于Dice系数[4]计算遗传距离,使用非加权组平均法(UPGMA)及Free Tree Version 0.9.1.50软件进行聚类分析,其中bootstrap值设置为1000,聚类图的修改使用Tree view 1.6.6软件;基于Dice系数的遗传距离使用NTSYS-pc Version 2.1软件进行主坐标分析。

表2 本研究中S-SAP分子标记引物组合信息表
注:M和E之后的小写字母代表选择性碱基。
2结果与分析
2.1S-SAP引物组合多态性分析
基于杨梅全基因组DNA序列,使用LTR_STRUC version 1.1软件分析预测逆转座子LTR位点,在预测结果中选择3个LTR区域相似度大于99%且长度大于150 bp的Ty1-copia逆转座子进行引物设计,共设计24个引物组合用于选择性扩增,随机选择8个杨梅品种进行引物组合的预筛选及有效性鉴定。结果发现:18个引物组合成功扩增,占总引物的75%;然后使用48份杨梅试材对这些引物组合进行多态性评价,结果共扩增出3739个100~500 bp的条带,平均每个引物可扩增208个条带(表3)。此外,平均有效等位基因位点数量和Shannon信息指数分别为1.3750和0.3599。每个引物组合扩增条带数量范围为3~193,其中,引物组合JY20的多态性最高,可扩增出292个条带,Shannon信息指数为0.4481。
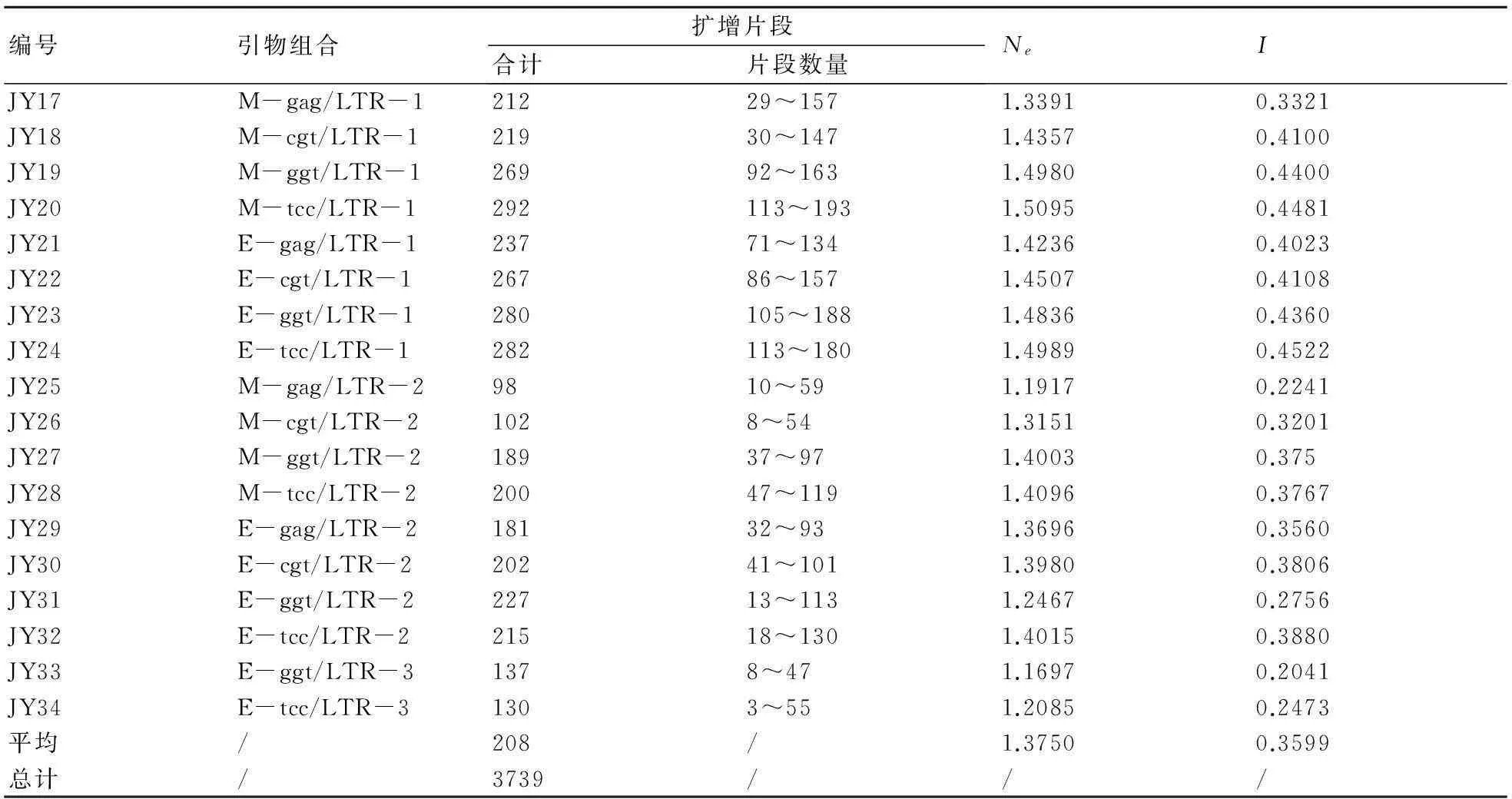
表3 S-SAP引物组合特征信息表
注:Ne代表有效等位基因数;I代表Shannon遗传多样性指数。
2.2杨梅种质资源遗传多样性分析
基于18个S-SAP引物组合并使用UPGMA构建48份试材的遗传亲缘关系聚类树(图1),共分为3个组及3个亚组。A组包含有A-1、A-2、A-3亚组。A-1亚组包括19份试材,包括荸荠和白杨梅等主栽品种。J2015-9、J2015-10、J2015-11为早熟优株,它们与荸荠品种划分于同一分支,意味着它们可能与荸荠品种的亲缘关系比较近。A-2亚组包括有9份试材,主要为大果类型品种,例如东魁、瑞光及来自浙江丽水的J2015-6等。A-3亚组主要是来自广东的杨梅品种。B组包括有12份试材,其中包括来自江苏的著名品种大浮大叶细蒂和大浮小叶细蒂。而C组品种均来自于江苏。
基于Nei等[4]遗传距离系数进行主坐标分析,前3个主坐标的方差贡献率分别为8.7%、7.4%和5.0%(图2-Ⅰ),通过比较图1和图2可以看出,系统聚类分析和主坐标分析所得结果基本上一致。但是值得注意的是,图2-Ⅰ中A-3亚组的27号样品在聚类分析(图1)及图2-Ⅱ中均被划分于B组;另外,其B组中的4和15号样品在聚类分析(图1)以及三维主坐标分析(图2-Ⅱ)均划分于C组,除此以外,其他品种的分类结果一致,由此可知,尽管聚类分析与三维主坐标分析结果完全一致,但是后者能够进一步从不同方向、不同层次展示各品种间的亲缘关系。

图1 48份杨梅试材UPGMA聚类树

Ⅰ.二维散点图;Ⅱ.三维散点图;A-1、A-2、A-3和
3讨论与结论
在前期研究中,一些DNA分子标记技术,例如AFLP、ISSR和SSR已经广泛应用于杨梅遗传多样性研究分析中[5-15]。然而,尽管大量的分子标记相继被开发,但是还不够全面且多态性普遍较低,在同等条件下用于鉴定同等数量植物品种及基因型则需要较高的实验成本。事实上,每种分子标记技术都是基于不同原理,但它们都或多或少可以反映来自基因组的变异[16];其中,S-SAP是一种基于基因组中普遍存在的、多拷贝、高度异质、插入位点多态的反转录转座子的分子标记,其技术体系比较成熟,也是多态性及灵敏度最高的分子标记之一,可以高效地进行植物品种鉴定,目前已在柑橘[16]、番茄[17]、莴苣[18]等植物上得到应用。本研究共获得18个S-SAP引物组合,平均每个引物组合可扩增获得208个条带,远高于前期研究中AFLP分子标记的多态性(39.3个条带)[12]、ISSR分子标记(4.9个条带)[6]、SSR分子标记多态性(8.3个条带)[5]。由此可见,S-SAP分子标记对于品种鉴定具有更高的多态性和检测效率。此外,基于前期的研究基础[3,19],在选择性扩增过程中加入一个嵌有荧光基团的通用引物M13,并在选择性扩增引物上游添加M13序列,该方法可以较大幅度降低实验成本,避免了使用传统聚丙烯酰胺凝胶电泳及银染等繁琐的操作方法,同时大幅度提高了检测分辨率,可以广泛应用于今后杨梅遗传多样性分析研究及基因组学的深入开展。
通过对48份杨梅试材的聚类分析发现,聚类结果与地域来源具有较为密切的关系,但是聚类分析以及主坐标分析结果均表明与果实色泽性状没有较为明显的联系,意味着杨梅果实色泽性状的形成具有较为复杂的遗传决定机制,具体原因还需要今后进一步探索研究,该结果与前期研究一致[5,9]。另外,聚类分析中B组中的大浮乌梅和马山白杨梅均为来自江苏的品种,它们聚在一个独立的分支且远离其他品种,由此可见,两者具有较为独特的遗传背景。同时,值得注意的是,图2-Ⅰ中A-3亚组的27号样品在主坐标三维散点图中(图2-Ⅱ)被划分于B组,事实上从后者图中可以看出,27号样品与B组样品的三维空间距离较小,说明它们之间的亲缘关系更为接近,故而将其划分于B组更能真实反映品种间的亲缘关系,该结果与聚类分析结果一致,然而,三维主坐标分析能提供更加全面且丰富的信息。因此,在基于相同遗传距离计算方法的条件下,可以优先选择采用三维主坐标分析的方法。
总之,本研究中所构建的S-SAP分子标记体系具有高效、稳定和便捷的特点;此外,利用该分子标记技术进行遗传多样性分析能够较为真实地反映品种之间的亲缘关系,因此,该技术可为今后在杨梅遗传多样性等方面研究提供有效的手段。
参考文献:
[1] Labra M, Imazio S, Grassi F, et al. Vine-1 retrotransposon-based sequence-specific amplified polymorphism forVitisviniferaL. genotyping[J]. Plant Breeding, 2004, 123(2): 180-185.
[2] 何平,李林光,李慧峰,等.S-SAP分子标记开发及其在苹果芽变鉴别上的应用[J].植物遗传资源学报,2013(2):298-302.
[3] Jiao Y, Ma R J, Shen Z J, et al. Development of Ty1-copiaretrotransposon-based S-SAP molecular markers for the study of genetic diversity in peach[J]. Biochem Syst Ecol, 2014, 57: 270-277.
[4] Nei M, Li W H. Mathematical model for studying genetic variation in terms of restriction endonucleases[J]. P Natl Acad Sci USA, 1979, 76(10): 5269-5273.
[5] Jiao Y, Jia H M, Li X W, et al. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myricarubra)[J]. BMC Genomics, 2012, 5(11): 151-154.
[6] Xie X B, Qiu Y Y, Ke L P, et al. Microsatellite primers in red bayberry,Myricarubra(Myricaceae)[J]. Am J Bot, 2011, 98(4): 93-95.
[7] Zhang S M, Gao Z S, Xu C J, et al. Genetic diversity of Chinese bayberry (MyricarubraSieb. et Zucc.) accessions revealed by Amplified Fragment Length Polymorphism[J]. Hortscience, 2009, 44(2): 487-491.
[8] Zhang S Y, Li X, Feng C, et al. Development and characterization of 109 polymorphic EST-SSRs derived from the Chinese bayberry (Myricarubra, Myricaceae) transcriptome[J]. Am J Bot, 2012, 99(12): 501-507.
[9] Xie R J, Zhou J, Wang G Y, et al. Cultivar Identification and genetic diversity of Chinese bayberry (Myricarubra) accessions based on fluorescent SSR markers[J]. Plant Mol Biol Rep, 2011, 29(3): 554-562.
[10] Zhang S, Xu C, Gao Z, et al. Development and characterization of microsatellite markers for Chinese bayberry (MyricarubraSieb. & Zucc.)[J]. Conserv Genet, 2009, 10(5): 1605-1607.
[11] Terakawa M, Kikuchi S, Kanetani S, et al. Characterization of 13 polymorphic microsatellite loci for an evergreen tree,Myricarubra[J]. Mol Ecol Notes, 2006, 6(3): 709-711.
[12] 张水明.基于AFLP和SSR分子标记的中国杨梅遗传多样性分析[D].杭州:浙江大学,2009.
[13] 郑金土,张望舒,鲍露,等.荸荠种杨梅变异新株系甬选56号的主要生物学特征及其AFLP鉴别[J].果树学报,2006(3):397-400.
[14] Jia H M, Shen Y T, Jiao Y, et al. Development of 107 SSR markers from whole genome shotgun sequences of Chinese bayberry (Myricarubra) and their application in seedling identification[J]. J Zhejiang Univ Sci B, 2014, 15(11): 997-1005.
[15] Jia H M, Jiao Y, Wang G Y, et al. Genetic diversity of male and female Chinese bayberry (Myricarubra) populations and identification of sex-associated markers[J]. BMC Genomics, 2015, 16(1): 394.
[16] Biswas M K, Chai l J, Amar M H, et al. Comparative analysis of genetic diversity inCitrusgermplasm collection using AFLP, S-SAP, SAMPL and SSR markers[J]. Scientia Horticulturae, 2011, 129(4): 798-803.
[17] Tam S M, Mhiri C, Vogelaar A, et al. Comparative analyses of genetic diversities within tomato and pepper collections detected by retrotransposon-based S-SAP, AFLP and SSR[J]. Theor Appl Genet, 2005, 110(5): 819-831.
[18] Pasquali M, Saravanakumar D, Gullino M L, et al. Sequence-specific amplified polymorphism (S-SAP) technique to analyseFusariumoxysporumf. sp. lactucae VCG 0300 isolate from lettuce[J]. J Plant Pahtology, 2008, 90(3): 527-535.
[19] Schuelke M. An economic method for the fluorescent labeling of PCR fragments[J]. Nat biotechnol, 2000, 18(2): 233-234.
(责任编辑:曾小军)
Establishment and Application of S-SAP Molecular Marker System in Chinese Bayberry (Myricarubra)
JIAO Yun, SHU Qiao-yun, LIU Zhu-qin, ZHANG Jian-hong
(Institute of Forestry, Ningbo Academy of Agricultural Science, Ningbo 315040, China)
Abstract:Designed S-SAP markers based on the LTR (long terminal repeat) sequences of Ty1-copia retrotransposons, used them for DNA profiling of 48 individuals of Chinese bayberry, evaluated the index of polymorphism, constructed a distance tree using clustering with UPGMA, and two-dimensional and three-dimensional plot principal coordinate analysis (PCO) were performed to analyze the genetic relationship of the Chinese bayberry samples. Successfully obtained amplicons from 18 primer combinations, analyzed yielded an average of 208 fragments per primer combination, adding up to a total of 3739 polymorphic fragments whose sizes ranged from 100 to 500 base pairs (bp). The number of markers for each genotype ranged from 98 to 292, and Shannon’s information index ranged from 0.2041~0.4522. The primer combination JY20 generated a total of 292 bands and was the most polymorphic. Reconstructed a UPGMA tree based on the above-mentioned 18 S-SAP markers, which were further divided into three groups and three subgroups. The cluster analysis and principal coordinate analysis results showed that the accessions in the same geographical region were more closely related than those from different regions, and which did not correlate with fruit color, using the three-dimensional plot of the PCO analysis could be more fully and more truly explain the genetic relationships of samples.
Key words:Chinese bayberry; S-SAP; Molecular markers; Genetic diversity
收稿日期:2015-09-24
基金项目:宁波市农业科学研究院院长基金(2015NKYP001)。
作者简介:焦云(1983─),男,河北石家庄人,副研究员,博士,研究方向:果树遗传育种与栽培技术。
中图分类号:S667.6
文献标志码:A
文章编号:1001-8581(2016)04-0001-06
