青蒿素对实验性自身免疫性重症肌无力大鼠R97-116抗体及细胞因子的影响
2016-06-17王艳君孟庆芳王思李衍滨
王艳君 孟庆芳 王思 李衍滨
261053 潍坊医学院(王艳君、王思);250016山东中医药大学第二附属医院(孟庆芳);250014山东大学附属千佛山医院(李衍滨)
青蒿素对实验性自身免疫性重症肌无力大鼠R97-116抗体及细胞因子的影响
王艳君孟庆芳王思李衍滨
261053 潍坊医学院(王艳君、王思);250016山东中医药大学第二附属医院(孟庆芳);250014山东大学附属千佛山医院(李衍滨)
摘要:目的探讨青蒿素(artemisinin)对实验性自身免疫性重症肌无力(EAMG)大鼠R97-116抗体及干扰素γ(IFN-γ)、白细胞介素17(IL-17)表达水平的影响。方法采用鼠源AChRα亚基97-116肽段免疫方法建立EAMG大鼠模型,将造模成功的大鼠20只随机分为青蒿素小、中、大剂量组和EAMG对照组。青蒿素小、中、大剂量组分别给予15、30、45 mg/(kg·d)青蒿素溶液灌胃治疗,1次/d,模型对照组给予等浓度二甲基亚砜水溶液灌胃。评测各组大鼠体质量和临床症状,采用流式细胞术检测淋巴结单个核细胞悬液细胞因子IFN-γ、IL-17水平,ELISA法检测血清抗R97-116 IgG/IgG1/IgG2b水平。结果青蒿素中、高剂量组大鼠体质量高于对照组(P<0.05);青蒿素各剂量组大鼠临床评分低于对照组(P<0.05)。青蒿素各治疗组IFN-γ、IL-17水平均低于对照组(P<0.01)。青蒿素15 mg/(kg·d)剂量组血清IgG、IgG1、IgG2b水平与对照组比较差异无统计学意义(P>0.05);30 mg/(kg·d)剂量组血清IgG(P<0.05)、IgG1(P<0.01)、IgG2b(P<0.01)水平低于对照组;45 mg/(kg·d)剂量组血清IgG(P<0.05)、IgG1(P<0.01)水平低于对照组。结论青蒿素能改善EAMG大鼠临床症状,对EAMG大鼠具有免疫调节作用,其机制可能与其通过直接或间接降低血清抗R97-116抗体水平、抑制淋巴结单个核细胞分泌IFN-γ和IL-17促炎性因子有关。
关键词:青蒿素;重症肌无力,实验性,自身免疫性;抗R97-116抗体;干扰素Ⅱ型;白细胞介素17
重症肌无力(myasthenia gravis,MG)是抗体介导、细胞免疫依赖、补体参与的、累及神经-肌肉接头(neuromuscular junction,NMJ)处的器官特异性自身免疫性疾病[1]。其主要发病机制为激活的乙酰胆碱受体(acetylcholine receptor,AChR)特异性T细胞表达和分泌多种细胞因子,并辅助B细胞产生AChR抗体,抗体与突触后膜AChR结合并激活补体系统,导致在NMJ处突触前膜产生的神经系统兴奋性递质传递障碍[2]。实验性自身免疫性重症肌无力(experimental autoimmune myasthenia gravis,EAMG)是研究人类MG的理想动物模型,其临床及免疫病理学表现、电生理学现象与人类MG颇为相似[3]。青蒿素作为含有过氧基的倍半帖内酯化合物,抗疟治疗效果显著。近年研究发现,青蒿素类药物具有免疫抑制、抗炎、诱导细胞凋亡、抑制血管增生等[4-7]药理作用,且己被用于研究治疗实验性自身免疫性脑脊髓炎、胶原性关节炎等各种自身免疫性疾病动物模型[8-9]。该研究采用R97-116肽段免疫Lewis大鼠制备EAMG模型,观察青蒿素对EAMG大鼠的免疫调节作用,旨在为青蒿素治疗MG提供客观的科学依据。
1材料和方法
1.1实验动物健康雌性Lewis大鼠23只,6~8周龄,体质量150 g左右,购自北京维通利华实验动物技术有限公司,在SPF环境下饲养。
1.2主要试剂鼠源AChRα亚基97-116 肽段序列(DGDFAIVKFTKVLLDYTGHI)由西安联美生物科技有限公司合成;青蒿素由天津化标生物技术有限公司提供;MEM、二甲基亚砜均购自美国Sigma-Aldrich 公司;胎牛血清(FBS)购自美国Thermo Fisher;生物素标记兔抗大鼠IgG购自北京博奥森生物技术有限公司;Biotin Anti-rat IgG1、IgG2b购自美国 Biolegend公司;小鼠抗大鼠IL-17A抗体购自美国eBioscience公司;小鼠抗大鼠干扰素γ(IFN-γ)抗体购自美国BD Pharmingen。
1.3方法
1.3.1EAMG模型诱导及评价:采用鼠源AChR-α97-116肽段二次免疫的方法建立EAMG 动物模型[10-11]。采用双盲评估法,自免疫第0天开始隔天评测大鼠体质量和临床症状,至免疫后第43天。临床评分采用Baggi等标准[11],包括震颤、驼背姿势、肌肉力量以及疲劳性(疲劳试验,即让大鼠重复抓握笼顶30 s再测量)。肌力具体分级标准为:0 级:正常肌力;1级:轻度活动减少,抓握力或叫声减弱,尤其在重复抓握后更加明显;2级:重复抓握前即出现震颤、低头、隆背、抓握力弱;3级:在抓握前即有严重的肌无力表现,无力抓握,濒死状态;4级:死亡。症状介于2 个级别之间者,取其平均值。造模成功标准:出现体质量下降或增长缓慢、肌肉震颤、垂首驼背姿势、前肢肌无力、活动减少且易疲劳症状。最终造模成功大鼠为20只。
1.3.2给药剂量与方法:将造模成功的20只大鼠随机分为青蒿素小、中、大剂量组和EAMG对照组,每组5只。于初次免疫后第6天,即EAMG大鼠发病初期,分别按体重15、30、45 mg/(kg·d)给予青蒿素各剂量组大鼠青蒿素水溶液(以二甲基亚砜为溶剂配置)灌胃,1次/d,直至第43天(发病高峰期);对照组大鼠给予等浓度二甲基亚砜水溶液。
1.3.3血清抗R97-116IgG/IgG1/IgG2b抗体水平测定:首次免疫后第43天处死大鼠,留取心脏血血清-20℃保存待测。采用ELISA法测定血清抗R97-116抗体水平。将R97-116(5 μg/mL)100 μL/孔包被96孔板,4℃过夜。弃包被液,用含0.05%(体积分数)Tween20的PBS洗板,然后加入含10%(体积分数)FBS的封闭液200 μL/孔,室温1.5 h;弃液加入稀释血清(1∶100),100 μL/孔,每例标本做平行实验3份,室温2 h;洗板后加入相应抗体:IgG(1∶3000)、IgG1(1∶1000)、IgG2b(1∶500),100 μL/孔,37 ℃孵育1 h;洗板后加入HRP标记的链霉亲和素(1∶1000);37 ℃孵育30 min后洗板,加入TMB 显色10 min;然后加入100 μL/孔终止液,采用酶标仪测定450 nm波长的吸光度值。
1.3.4细胞因子检测:首次免疫后第43天处死大鼠,无菌条件下取双侧腹股沟淋巴结,在盛有滤网的培养皿中用无菌针栓研磨淋巴结,制备淋巴结单个核细胞(mononuclear cells,MNC)悬液。细胞悬浮于含有1%(体积分数)MEM、50 μg/mL庆大霉素和10%(体积分数)FBS培养基中,调整细胞水平为2×106个/mL。采用流式细胞术检测淋巴结MNC悬液IFN-γ和白细胞介素-17(IL-17)水平。每管取上述淋巴结MNC悬液1×106个/mL,依次加入2%(质量浓度)多聚甲醛固定细胞、破膜工作液破膜,洗涤后避光加入FITC标记的小鼠抗大鼠IFN-γ抗体(或PE标记的小鼠抗大鼠IL-17抗体),4℃孵育30 min,细胞重悬过滤后上机检测。
1.4统计学处理采用SPSS17.0统计软件分析,计量资料以均数±标准差表示,符合正态分布且方差齐性的数据采用单因素方差分析,两两比较用LSD-t检验;不满足方差齐性者采用Kruskal-Wallis(K-W)检验,两两比较采用Mann-WhitneyU检验。以P<0.05表示差异具有统计学意义。
2结果
2.1大鼠体质量和临床症状评分与对照组比较,青蒿素15 mg/(kg·d)剂量组体质量变化无统计学差异(P>0.05);30 mg/(kg·d)剂量组免疫后第12~18、30~38及42天体质量增高(均P<0.05);45 mg/(kg·d)剂量组免疫后第12~18、34~38天体质量增高(均P<0.05)。青蒿素各剂量组间体质量比较差异无统计学意义(P>0.05)。青蒿素15 mg/(kg·d)剂量组在免疫后第20~43天临床症状评分低于对照组(P<0.05);30 mg/(kg·d)剂量组在第16~43天临床评分低于对照组(P<0.05);45 mg/(kg·d)剂量组在免疫后第14~43天临床症状评分低于对照组(P<0.05)。青蒿素各剂量组之间大鼠临床评分比较差异无统计学意义(P>0.05)。具体结果见图1、2。

EAMG:实验性自身免疫性重症肌无力,图2~4同;青蒿素30 mg/(kg·d)组与对照组分别比较,aP<0.05;青蒿素45 mg/(kg·d)组与对照组比较,bP<0.05 图1 各组EAMG大鼠免疫后不同时间体质量变化趋势

与青蒿素45 mg/(kg·d)组比较,aP<0.05;与青蒿素30 mg/(kg·d)组比较,bP<0.05;与青蒿素各剂量组分别比较,cP<0.05 图2 各组EAMG大鼠免疫后不同时间临床评分变化
2.2大鼠血清R97-116 抗体水平检测青蒿素15 mg/(kg·d)剂量组血清IgG、IgG1、IgG2b水平与对照组比较差异无统计学意义(P>0.05);30、45 mg/(kg·d)剂量组血清IgG(P<0.05)、IgG1(P<0.01)、IgG2b(P<0.01)水平均低于对照组;45 mg/(kg·d)剂量组血清IgG(P<0.05)、IgG1(P<0.01)水平均低于对照组。青蒿素各组间比较血清IgG水平差异无统计学意义(P>0.05);30 mg/(kg·d)剂量组血清IgG1水平均低于15 mg/(kg·d)剂量组(P<0.01,P<0.05),30 mg/(kg·d)剂量组。血清IgG2b水平低于15 mg/(kg·d)剂量组(P<0.01)。结果见图3。
2.3大鼠淋巴结MNC悬液细胞因子水平青蒿素小、中、大剂量组淋巴结MNC悬液IFN-γ和IL-17水平均明显低于对照组(均P<0.01),但青蒿素各剂量组间比较差异无统计学意义(P>0.05)。具体结果见图4。
3讨论

aP<0.05;bP<0.01图3 不同剂量青蒿素对EAMG大鼠血清R97-116 抗体水平的影响
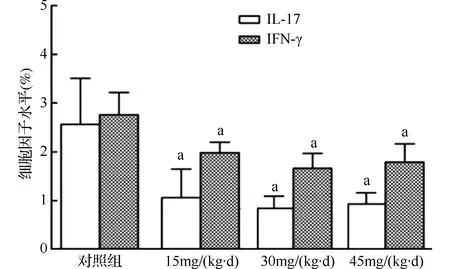
与对照组比较,aP<0.01图4 不同剂量青蒿素对EAMG大鼠淋巴结MNC悬液细胞因子水平影响
EAMG动物模型是研究MG发病机制和寻求有效治疗方法的理想模型。Baggi等[11]采用鼠源AChRα97-116肽段二次免疫法,成功建立了EAMG模型,R97-116肽段能够成功诱导大鼠抗AChR和抗R97-116抗体的产生。本研究亦采用R97-116肽段诱导大鼠EAMG模型,通过观察不同剂量青蒿素对大鼠体质量、临床症状评分、R97-116抗体水平及细胞因子IFN-γ和IL-17水平的影响,初步探讨了青蒿素在防治EAMG中的可能作用及其可能机制。
EAMG大鼠的体质量和临床症状评分是衡量机体免疫功能的初步指标。本研究结果显示,初次免疫后第2天,各组大鼠体质量出现一过性下降,经强化免疫后,EAMG模型组大鼠与青蒿素各剂量组相比体质量出现明显下降,且随时间推移体质量增长缓慢;初次免疫后约1周,各组大鼠相继表现出肌无力症状,且在强化免疫后EAMG模型组大鼠与青蒿素各剂量组相比肌无力症状明显加重,并伴有明显的活动度减少、易疲劳、身体震颤等临床症状。抗AChR抗体的产生最终会导致EAMG的发生发展。AChR抗体以IgG型为主,其中IgG1和IgG2可以高效地与NMJ处突出后膜上的AChR结合并激活补体最终导致肌无力的发生与发展[12-13]。该实验结果显示,青蒿素干预的EAMG大鼠血清中抗R97-116 IgG/IgG1/IgG2b抗体的水平明显低于对照组,与其临床表现一致。推测青蒿素缓解EAMG大鼠的临床症状与降低其血清R97-116 IgG/IgG1/IgG2b抗体水平可能有密切关系。
在MG/EAMG 免疫学发病机制中,Th1型细胞具有重要的致病作用,IFN-γ是Th1型细胞分泌的主要促炎性细胞因子,可以激活抗原提呈细胞进一步扩增活化AChR特异性T 细胞,可以诱导B 细胞成熟、辅助突触后膜AChR 抗体的产生并引起NMJ功能的失调[14]。Wang等将IFN-γ转基因定位于EAMG大鼠NMJ处或体外给予IFN-γ,均可加重大鼠临床症状[15]。而IFN-γ基因敲除鼠对EAMG 的发生具有耐受性[16]。该实验结果表明,青蒿素可明显下调淋巴结MNC分泌IFN-γ的水平,表明青蒿素能够通过抑制单核细胞抗原提呈和AChR特异性T细胞应答的能力,进而抑制抗原特异性免疫应答,缓解EAMG大鼠的临床症状。在自身免疫性疾病的过程中,Th17细胞通过分泌炎症介质IL-17诱导免疫应答。有研究发现,Th17和IL-17参与小鼠EAMG 的发病过程,并且起到加速疾病进展的作用[17-18]。IL-17还可以促进T细胞的启动活化与增殖、辅助AChR 抗体的产生[19-20]。该研究结果显示,青蒿素能够下调淋巴结MNC中IL-17的水平,且可减轻EAMG大鼠临床症状,提示青蒿素可能通过控制致炎细胞因子的释放、抑制T淋巴细胞介导的免疫反应、抑制B细胞的增殖和分化、减少抗体对NMJ突触后膜的AChR破坏,进而缓解EAMG临床症状。
目前有关青蒿素及其衍生物相关的毒理学研究也逐步引向深入,以探索其新的临床适应证。动物毒性实验研究发现[21],青蒿素衍生物在较高剂量下可对神经系统造成损害。青蒿素的神经毒性主要是听力损伤、共济失调和震颤,其机制可能与氧化应激和线粒体的损伤有关。
综上所述,青蒿素能够改善EAMG大鼠的临床症状,可能与其直接或间接降低血清R97-116抗体水平、抑制淋巴结MNC分泌IFN-γ和IL-17促炎性因子有关,提示青蒿素可能对EAMG大鼠具有免疫调节作用。
参考文献:
[1]Soltys J,Wu X.Complement regulatory protein Crry deficiency contributes to the antigen specific recall response in experimental autoimmune myasthenia gravis[J].J Inflamm(Lond),2012,9(1):20.
[2]Vincent A.Unravelling the pathogenesis of myasthenia gravis[J].Nat Rev Immunol,2002,2(10):797-804.
[3]Link H,Xiao BG.Rat models as tool to develop new immunotherapies[J].Immunol Rev,2001,184:117-128.
[4]Wang JX,Tang W,Shi LP,et al.Investigation of the immunosuppressive activity of artemether on T-cell activation and proliferation[J].Br J Pharmacol,2007,150(5):652-661.
[5]Wang JX,Hou LF,Yang Y,et al.SM905,an artemisinin derivative,inhibited NO and pro-inflammatory cytokine production by suppressing MAPK and NF-kappaB pathways in RAW 264.7 macrophages[J].Acta Pharmacol Sin,2009,30(10):1428-1435.
[6]Disbrow GL,Baege AC,Kierpiec KA,et al.Dihydroartemisinin is cytotoxic to papillomavirus-expressing epithelial cells in vitro and in vivo[J].Cancer Res,2005,65(23):10854-10861.
[7]Anfosso L,Efferth T,Albini A,et al.Microarray expression profiles of angiogenesis-related genes predict tumor cell response to artemisinins[J].Pharmacogenomics J,2006,6(4):269-278.
[8]Li X,Li TT,Zhang XH,et al.Artemisinin analogue SM934 ameliorates murine experimental autoimmune encephalomyelitis through enhancing the expansion and functions of regulatory T cell[J].PLoS One,2013,8(8):e74108.
[9]Li Y,Wang S,Wang Y,et al.Inhibitory effect of the antimalarial agent artesunate on collagen-induced arthritis in rats through nuclear factor kappa B and mitogen-activated protein kinase signaling pathway[J].Transl Res,2013,161(2):89-98.
[10]Li XL,Liu Y,Cao LL,et al.Atorvastatin-modified dendritic cells in vitro ameliorate experimental autoimmune myasthenia gravis by up-regulated Treg cells and shifted Th1/Th17 to Th2 cytokines[J].Mol Cell Neurosci,2013,56:85-95.
[11]Baggi F,Annoni A,Ubiali F,et al.Breakdown of tolerance to a self-peptide of acetylcholine receptor alpha-subunit induces experimental myasthenia gravis in rats[J].J Immunol,2004,172(4):2697-703.
[12]Hu B,Tian X,Huang H,et al.Expression of IL-21 in the peripheral blood of myasthenia gravis patients and its correlation with anti-AChR-Ab class switch[J].Zhongnan Daxue Xuebao Yixueban,2010,35(9):958-963.
[13]Saoudi A,Bernard I,Hoedemaekers A,et al.Experimental autoimmune myasthenia gravis may occur in the context of a polarized Th1- or Th2-type immune response in rats[J].J Immunol,1999,162(12):7189-97.
[14]Wang W,Ostlie NS,Conti-Fine BM,et al.The susceptibility to experimental myasthenia gravis of STAT6-/- and STAT4-/- BALB/c mice suggests a pathogenic role of Th1 cells[J].J Immunol,2004,172(1):97-103.
[15]Wang HB,Shi FD,Li H,et al.Role for interferon-gamma in rat strains with different susceptibility to experimental autoimmune myasthenia gravis[J].Clin Immunol,2000,95(2):156-162.
[16]Liu R,Hao J,Dayao CS,et al.T-bet deficiency decreases susceptibility to experimental myasthenia gravis[J].Exp Neurol,2009,220(2):366-373.
[17]Wang W,Milani M,Ostlie N,et al.C57BL/6 mice genetically deficient in IL-12/IL-23 and IFN-gamma are susceptible to experimental autoimmune myasthenia gravis,suggesting a pathogenic role of non-Th1 cells[J].J Immunol,2007,178(11):7072-7080.
[18]Mu L,Sun B,Kong Q,et al.Disequilibrium of T helper type 1,2 and 17 cells and regulatory T cells during the development of experimental autoimmune myasthenia gravis[J].Immunology,2009,128(1Suppl):e826-836.
[19]Aricha R,Mizrachi K,Fuchs S,et al.Blocking of IL-6 suppresses experimental autoimmune myasthenia gravis[J].J Autoimmun,2011,36(2):135-141.
[20]Barbosa RR,Silva SP,Silva SL,et al.Primary B-cell deficiencies reveal a link between human IL-17-producing CD4 T-cell homeostasis and B-cell differentiation[J].PLoS One,2011,6(8):e22848.
[21]Efferth T,Kaina B.Toxicity of the antimalarial artemisinin and its dervatives[J].Crit Rev Toxicol,2010,40(5):405-421.
(本文编辑:时秋宽)
Influence of artemisinin on the expression of R97-116 antibody and cytokines in Lewis rats with experimental auto immune myasthenia gravis
WANGYanjun,MENGQingfang,WANGSi,LIYanbin*.
*DepartmentofNeurology,QianfoshanHospitalAffiliatedtoShandongUniversity,Ji′nanShandong250014,ChinaCorresponding author:LI Yanbin,Email:lch1226@sohu.com
ABSTRACT:ObjectiveTo investigate the influence of artemisinin on the expression of R97-116 antibody and interferon-gamma(IFN-γ),interleukin-17(IL-17)in Lewis rats with experimental autoimmune myasthenia gravis(EAMG). MethodsEAMG models were established by subcutaneous injection of synthetic peptide R97-116 into both hind footpads of Lewis rats.Twenty rats were randomly divided into four groups:the artemisinin low,medium,high dose group and the control group(n=5 in each group).Artemisinin was administrated daily by oral gavage at a dose of 15mg/(kg·d),30 mg/(kg·d)and 45 mg/(kg·d),and the control group were administrated with dimethyl sulphoxide(DMSO)solution of the same concentration.Clinical symptoms and body weight were evaluated,the levels of intracellular cytokines IFN-γ and IL-17 in mononuclear cell(MNC)were analyzed by flow cytometry,anti-R97-116 IgG/IgG1/IgG2b antibody in the serum were detected by ELISA. ResultsThe rats′weights were significantly higher in the medium and high dose group of artemisinin than that of the control group(P<0.05).Each group of artemisinin exhibited lower clinical scores than the control group(P<0.05).The secretion of IFN-γ in each group of artemisinin were lower than the control group(P<0.01),and so did IL-17.The levels of R97-116 IgG/IgG1/IgG2b were not significantly different between the low dose group of artemisinin and the control group(P>0.05).The levels of serum R97-116 IgG,IgG1 and IgG2b in the medium dose group of artemisinin were lower than the control group(P<0.05,P<0.01,P<0.01,respectively).The levels of serum R97-116 IgG and IgG1 in the high dose group of artemisinin were lower than the control group(P<0.05,P<0.01,respectively).ConclusionsIt is suggested that artemisinin could ameliorate EAMG by decreasing the level of anti R97-116 antibody in serum,down-regulating production of cytokines IFN-γ and IL-17 in lymph node mononuclear cells.
Key words:artemisinin;myasthenia gravis,experimental,autoimmune;anti-R97-116 antibody;type Ⅱ interferon;interleukin 17
doi:10.3969/j.issn.1006-2963.2016.03.002
基金项目:山东省自然基金课题(编号:ZR2010HM068)
通讯作者:李衍滨,Email:lch1226@sohu.com
中图分类号:R746.1
文献标识码:A
文章编号:1006-2963(2016)03-0167-05
(收稿日期:2015-10-14)
